製品基本Q&A
製品基本Q&A
カンサイダス®(カスポファンギン酢酸塩)
製品情報
本剤の電子添文には、以下のとおり記載されています。
4. 効能又は効果
○真菌感染が疑われる発熱性好中球減少症
○カンジダ属又はアスペルギルス属による下記の真菌感染症
○食道カンジダ症
○侵襲性カンジダ症
○アスペルギルス症(侵襲性アスペルギルス症、慢性壊死性肺アスペルギルス症、肺アスペルギローマ)
5. 効能又は効果に関連する注意
〈真菌感染が疑われる発熱性好中球減少症〉
5.1 本剤は以下の3条件を満たす症例に投与すること。
・1回の検温で38℃以上の発熱、又は1時間以上持続する37.5℃以上の発熱
・好中球数が500/mm3未満の場合、又は1,000/mm3未満で500/mm3未満に減少することが予測される場合
・適切な抗菌薬投与を行っても解熱せず、抗真菌薬の投与が必要と考えられる場合
5.2 発熱性好中球減少症の患者への投与は、発熱性好中球減少症の治療に十分な経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ実施すること。
5.3 発熱性好中球減少症に投与する場合には、投与前に適切な培養検査等を行い、起炎菌を明らかにする努力を行うこと。起炎菌が判明した際には、本剤投与継続の必要性を検討すること。
〈侵襲性カンジダ症〉
5.4 カンジダ血症、腹腔内膿瘍、腹膜炎、胸腔内感染以外における検討は行われていない。[17.1.1、17.1.2 参照]
〈侵襲性アスペルギルス症〉
5.5 他の治療が無効あるいは忍容性に問題がある患者に本剤の使用を考慮すること。
真菌(アスペルギルス属及びカンジダ属)細胞壁の主要構成成分である1,3-β-D-グルカンの生合成を阻害します。なお、哺乳類の細胞は、1,3-β-D-グルカンを合成しません。
<引用>
電子添文
使用方法
本剤の電子添文には、以下のとおり記載されています。
6. 用法及び用量
〈成人〉
真菌感染が疑われる発熱性好中球減少症
通常、カスポファンギンとして投与初日に70mgを、投与2日目以降は50mgを1日1回投与する。本剤は約1時間かけて緩徐に点滴静注する。
カンジダ属又はアスペルギルス属による下記の真菌感染症
・食道カンジダ症
通常、カスポファンギンとして50mgを1日1回投与する。本剤は約1時間かけて緩徐に点滴静注する。
・侵襲性カンジダ症、アスペルギルス症
通常、カスポファンギンとして投与初日に70mgを、投与2日目以降は50mgを1日1回投与する。本剤は約1時間かけて緩徐に点滴静注する。
〈小児〉
真菌感染が疑われる発熱性好中球減少症、カンジダ属又はアスペルギルス属による食道カンジダ症、侵襲性カンジダ症、アスペルギルス症
通常、カスポファンギンとして投与初日に70mg/m2(体表面積)を、投与2日目以降は50mg/m2(体表面積)を1日1回投与する。本剤は約1時間かけて緩徐に点滴静注する。なお、1日1回50mg/m2(体表面積)の投与で効果不十分の場合には、1日1回70mg/m2(体表面積)まで増量することができる。いずれの場合も1日用量として70mgを超えないこと。
7. 用法及び用量に関連する注意
〈成人〉
中等度の肝機能障害を伴う患者に対しては、下表を目安に本剤の用量調節をすること。[16.6.1 参照]
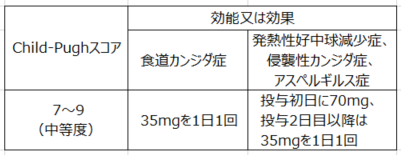
軽度の肝機能障害(Child-Pughスコア5~6)を伴う患者に対しては通常の用量を投与する。
重度の肝機能障害(Child-Pughスコア10以上)を伴う患者に対しては本剤の投与経験がない。
7.2 エファビレンツ、ネビラピン、リファンピシン、デキサメタゾン、フェニトイン、カルバマゼピンと本剤を併用する場合、本剤70mgの1日1回投与を検討すること。[10.2、16.7.3、16.7.4 参照]
〈小児〉
7.3 3ヵ月未満の患者では血中濃度が高くなる可能性があるので、3ヵ月未満の患者に投与する際は減量を考慮すること。[16.1.2 参照]
7.4 小児の肝機能障害患者に対する検討は行われていない。
7.5 エファビレンツ、ネビラピン、リファンピシン、デキサメタゾン、フェニトイン、カルバマゼピンと本剤を併用する場合、本剤70mg/m2(体表面積)の1日1回投与を検討すること。なお、1日用量として70mgを超えないこと。[10.2、16.7.3、16.7.4 参照]
電子添文上 「9.特定の背景を有する患者に関する注意」 に、高齢者に対する注意は記載されていません。
高齢者における本剤の用量調節は不要ですが、一般に高齢者では生理機能が低下しているので、注意してください。
本剤の電子添文には、以下のとおり記載されています。
9. 特定の背景を有する患者に関する注意
9.5 妊婦
妊婦又は妊娠している可能性のある女性には治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
ラットでは母動物に毒性があらわれる用量(5mg/kg/日)で、胎児体重の減少並びに頭蓋及び体躯の不完全骨化発現率の増加が認められている。さらに、同用量で頸肋の発現率増加がみられている。動物試験(ラット、ウサギ)で、胎盤通過が認められている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトの母乳中に移行するか否かは不明である。ラットでは乳汁移行が認められている。
本剤の電子添文には、以下のとおり記載されています。
9. 特定の背景を有する患者に関する注意
9.7 小児等
投与に際しては観察を十分に行うこと。小児の臨床試験では、成人と比べALT増加、AST増加、肝機能異常の発現頻度が高いことが報告されている。低出生体重児、新生児及び3ヵ月未満の乳児を対象とした国内臨床試験は実施していない。
外国第Ⅰ相試験で、軽度及び中等度肝機能障害患者における薬物動態、安全性を検討したデータに基づき、中等度肝機能障害成人患者では維持用量を35mg1日1回投与に減量するとしました。
<外国第Ⅰ相試験の詳細>
外国第Ⅰ相試験(009及び030試験)において、軽度肝機能障害患者(Child-Pughスコア:5~6)にカンサイダスⓇ 70mg を単回投与したとき、健康被験者と比較して血漿中薬物濃度(AUC0-∞ )の約55%の増加が認められ、また、投与初日に負荷用量70mg を投与後、第2~14日は維持用量として50mgを1日1回投与(以下、本剤70/50mg)したとき、第7日及び第14日の血漿中薬物濃度は健康被験者と比較してわずかな増加が認められました(AUC0-24hr が21~26%増加)。
中等度肝機能障害患者(Child-Pughスコア:7~9)にカンサイダスⓇ70mgを単回投与したとき、平均血漿中薬物濃度(AUC0-∞ )は健康被験者と比較して76%の増加が認められ、また、投与初日に負荷用量70mgを投与後、第2~14日は維持用量として35mgを1日1回投与したとき、第7日及び第14日の血漿中薬物濃度(AUC0-24hr )が健康被験者に本剤70/50mgを14日間反復投与したときの血漿中薬物濃度(AUC0-24hr )と類似していました(1)。
また、これらの肝機能障害患者を対象とした薬物動態試験では、重篤な副作用及び投与中止を要する副作用は認められませんでした。
上記の薬物動態データ及び安全性データに基づき、中等度肝機能障害成人患者では維持用量を本剤35mgの1日1回投与に減量することを、電子添文「7. 用法及び用量に関連する注意」の項に記載しています。
<引用>
(1)Mistry GC et al. J Clin Pharmacol. 2007;47(8):951-961.
国内臨床試験は、海外臨床試験の点滴静注の設定を根拠として1時間で実施し、この条件下で安全性が確認されているため、同様の投与時間で承認されました。
また、非臨床試験では、投与速度とヒスタミン遊離作用に関連が認められたため、臨床試験は約1時間かけて緩徐に点滴静注することと設定されています。
電子添文の禁忌に記載がなく、投与可能と考えられます。
<透析患者>
米国の添付文書には「投与に際して、カスポファンギンは透析により除去されないため、血液透析後の補足投与の必要はない」と記載され(1)、FDAに承認されています。
米国添付文書には腎機能障害に関して以下のように記載されています(1)。
「70 mg 単回投与の臨床試験では、軽度腎機能障害患者(クレアチニン・クリアランス:50~80 mL/分)の薬物動態は腎機能正常被験者と類似していた。中等度(クレアチニン・クリアランス:31~49 mL/分)、重度(クレアチニン・クリアランス:5~30 mL/分)、及び末期(クレアチニン・クリアランス:10 mL/分未満で透析中)の腎機能障害患者では、単回投与後のカスポファンギンの血漿中薬物濃度がやや増加した(AUC範囲:30~49%)。しかし、侵襲性アスペルギルス症及びカンジダ血症又はその他のカンジダ感染(腹腔内膿瘍、腹膜炎、胸膜腔内感染)の成人患者に本剤50mg を1日1回反復投与した際、軽度から末期の腎機能障害によるカスポファンギン薬物濃度への意味のある影響は認められなかった。したがって、腎機能障害患者への用量調整は必要ない。カスポファンギンは透析により除去されないため、血液透析後の補足投与の必要はない。」
<腎機能障害患者>
腎機能障害患者への投与において、減量は必要ないと判断されています。
海外での腎機能障害患者の薬物動態データに基づく母集団解析結果から、腎機能は血中濃度(C1hr 、C24hr)に影響を与える因子ではないことが示されたためです(2)。
<引用>
(1)米国添付文書
(2)インタビューフォーム Ⅶ.薬物動態に関する項目 10.特定の背景を有する患者 1)腎機能障害患者(外国人データ)
中心静脈栄養(TPN(*))を再開する場合、配合変化が生じる可能性が否定できないため、フラッシュすることを推奨しています。
(*)TPN(Total Parenteral Nutrition):中心静脈栄養
安全性
〈成人〉
主な副作用として、AST増加、ALT増加、高血圧、好酸球数増加、悪心、静脈炎、血中Al-P増加、血中カリウム減少、γ-GTP増加、プロトロンビン時間延長などが報告されています。
〈小児〉
主な副作用として、ALT増加、AST増加、肝機能異常、LDH増加、γ-GTP増加などが報告されています。
<引用>
電子添文
その他
調製後は速やかに使用してください。やむを得ず保存を必要とする場合でも、バイアル中で溶解した本剤の溶液は、25℃以下で24時間以内に使用してください。また、希釈した点滴静注液は、25℃以下では24時間以内、冷所(2~8℃)では48時間以内に使用してください。
<引用>
電子添文
貯法は、2~8℃です。
<引用>
電子添文
<アスペルギルス症に対する有効性>

043試験:小児患者対象 海外第Ⅱ相試験
062試験:成人患者対象 国内第Ⅲ相試験
019試験:成人患者対象 海外第Ⅱ相試験
ブドウ糖含む希釈液を使用した安定性試験において、含量低下が認められましたが、具体的な数値については開示していません。また、臨床においてのデータもありません。
ブドウ糖を含む希釈液は使用を避けてください。
非臨床試験の結果を踏まえて設定されています(1)。
ラット及びサルの反復投与毒性試験において、無毒性量を超える用量で投与部位に刺激性変化(主な所見として、細胞浸潤、線維成分増加、出血、血管周囲の壊死あるいは血栓症等)が認められ、この投与局所の変化は投与液の濃度と投与速度に依存すると考えられました。
この結果を踏まえ、臨床試験でのカスポファンギンの濃度は、1試験の1用量を除き、0.5mg/mL以下と規定して実施し、局所忍容性が全般的に良好であったため臨床用量においても最終濃度は0.5mg/mLを超えないこととされました。
<引用>
(1) 審査報告書(2012年01月18日) p.58
調製によるバイアルや注射器内の損失を考慮したためです。
10.5mLで溶解し、10mLを抜き取ることで、電子添文の用法及び用量を下回らない、十分な投与量を確保できます。
成人の場合、電子添文に記載されているように0.5mg/mLを超えない濃度にするためには、計算上、溶解した液を下表の「希釈液量」以上の生理食塩液に希釈する必要があります。
<溶解>
本剤1バイアル(70mgバイアル又は50mgバイアル)に、生理食塩液あるいは注射用水10.5mLを注入し、ゆっくりと振り混ぜて完全に溶解させます。溶解後の濃度は、7.2mg/mL(70mgバイアル)又は5.2mg/mL(50mgバイアル)になります。
<希釈>
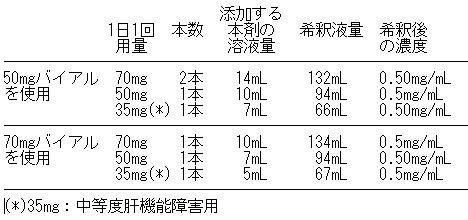
50mg、70mgのバイアルをそれぞれ10.5mLに溶解し、溶液10mLを点滴バッグに注入すると、計算上の用量はそれぞれ52mg、72mgとなります。
電子添文記載の投与量である、50mg又は70mgを確実に投与できることを考慮しました。
また、調製時のバイアルや注射器内の損失などを考慮して、電子添文の用法及び用量を下回らない、十分な投与量を確保できるように設定されています。
臨床試験(治験)も同様の調製方法で実施されています。
国内、海外で実施された臨床試験において、投与時の薬液濃度が0.525mg/mLを超えた場合のデータがないため、70mg投与時にもこれを超えない用量として250mLが選択されました。
調整後の最終濃度が0.5mg/mLを超えない範囲で、希釈液量を減じることは可能です。
インタビューフォーム「Ⅳ.製剤に関する項目 7.調製法及び溶解後の安定性 表Ⅳ-5 点滴静注液の調製法」をご参照ください。
電子添文[調製後の最終濃度が0.5mg/mLを超えないこと。]
70mgを100mLで希釈する調製法は推奨していません。
調製後の最終濃度が0.5mg/mLを超えないようにしてください。
各試験背景が異なるものの海外の治験結果を集積した報告では、各菌種に対して有効と判断された症例の割合に顕著な違いはありませんでした(1)。
| C. glabrata | 85%(46/54) |
| C. guilliermondii | 89%(8/9) |
| C. krusei | 70%(7/10) |
| C. lusitaniae | 100%(5/5) |
| C. parapsilosis | 74%(52/70) |
| C. tropicalis | 71%(46/65) |
| C. albicans | 7%(127/166) |
<引用>
(1)Colombo AL et al. Antimicrob Agents Chemother. 2010;54(5):1864-1871.