国際共同第Ⅲ相試験:KEYNOTE-A18試験
子宮頸癌:国際共同臨床試験成績
国際共同第Ⅲ相試験<KEYNOTE-A18試験>
1)承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-A18試験)
2)Lorusso D et al. Lancet 2024; 403: 1341-1350
3)Lorusso D et al. Lancet 2024; 404: 1321-1332
本試験はMSD社の資金提供により行われた。著者のうち、Kan Li、Karin Yamada、Sarper Toker、Stephen M Keefe、Peng Liuは同社の社員である。
試験概要
【目的】
未治療*1の局所進行子宮頸癌患者におけるキイトルーダ®と同時化学放射線療法(CCRT:シスプラチン同時併用下での外部照射、及びその後の小線源治療)との併用群(以下、キイトルーダ®+CCRT群)と、プラセボとCCRTとの併用群(以下、プラセボ+CCRT群)の有効性及び安全性を比較検討する。
【デザイン】
国際共同無作為化二重盲検第Ⅲ相試験[優越性試験](第1回中間解析データカットオフ日:2023年1月9日)(第2回中間解析データカットオフ日:2024年1月8日)
【対象】
未治療のFIGO 2014進行期分類のⅠB2~ⅡB期*2(リンパ節転移陽性)又はⅢ~ⅣA期*3(リンパ節転移陽性又は陰性)の局所進行子宮頸癌患者*41,060例(日本人90例を含む)
【方法】
キイトルーダ®+CCRT群[キイトルーダ®200mgをQ3Wで5サイクル、シスプラチン40mg/m2をQWで5回又は6回投与、及び放射線治療(外部照射とその後の小線源治療)*5後、キイトルーダ®400mgをQ6Wで15サイクル投与]又はプラセボ+CCRT群[プラセボをQ3Wで5サイクル及びキイトルーダ®+CCRT群と同一の化学放射線療法後、プラセボをQ6Wで15サイクル投与]に1:1の割合で無作為に割り付け、投与完了又は疾患進行等による投与中止まで継続した。最初の画像評価は、CCRTの完了から12週間後に実施した。2年目までは12週間間隔、3年目は24週間間隔、それ以降は1年間隔で実施した。
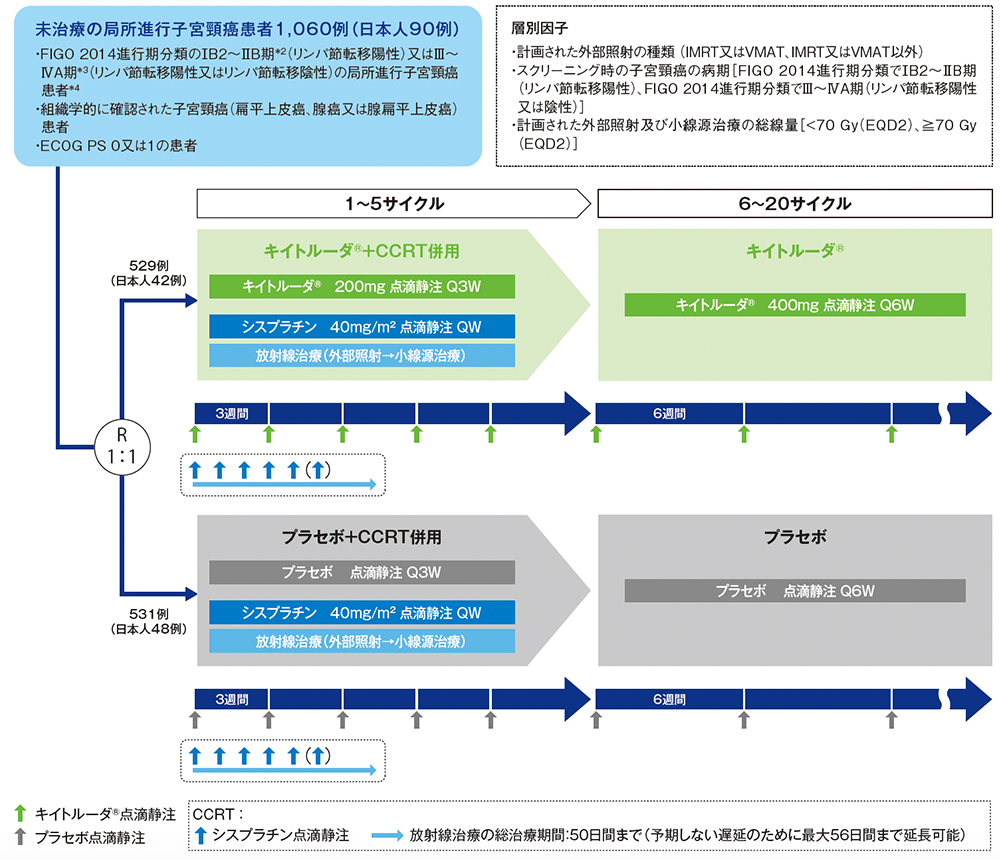
シスプラチンは、5回又は6回投与とされた。キイトルーダ®又はプラセボ及びシスプラチン両剤の投与日は、シスプラチンの投与前にキイトルーダ®又はプラセボを投与することが望ましい(治験実施医療機関の標準手順に従いシスプラチンをキイトルーダ®又はプラセボの前に投与することも可能)。
シスプラチンの投与は、外部照射の施行予定日の放射線治療前に行うこととされたが、日本では、標準的な手順であれば外部照射の後にシスプラチンを投与することが許容された。外部照射は、各国の標準的な手順に従い、中央遮蔽を適用してもよい。
FIGO:国際産婦人科連合 IMRT:強度変調放射線治療 VMAT:強度変調回転放射線治療 EQD2:2Gy換算等価線量 Q3W:3週間間隔 Q6W:6週間間隔 QW:1週間間隔
【評価項目】
主要評価項目:無増悪生存期間(progression free survival:PFS)a※及び全生存期間(overall survival:OS)※
副次評価項目: 2年PFS率a、3年OS率、CCRT実施後12週時点の完全奏効(CR)率b、奏効率(overall response rate:ORR)b、安全性など
探索的評価項目:奏効期間(duration of response:DOR)bなど
※検証的解析項目
【解析計画】
解析対象集団:主な有効性の解析はITT*6集団、ORR、CR率、DORの解析は、無作為化され、治験組入れ時に測定可能病変を有する患者集団、安全性の解析はAPaT*7集団を対象として実施した。
有効性評価の統計手法: PFS、OS、2年PFS率、3年OS率はKaplan-Meier法を用いて推定した。PFS、OSの群間比較には、層別ログランク検定を用い、投与群を共変量とした層別Cox比例ハザードモデルを用いて、ハザード比(HR)及び95%信頼区間(95%CI)を算出した。ORRの群間比較には層の例数を重みとする層別Miettinen and Nurminen法を用いた。
層別ログランク検定、層別Cox比例ハザードモデル及び層別Miettinen and Nurminen法の層別因子には、無作為化層別因子*8を用いた。PFS及びOSについて、投与群を共変量としたCox比例ハザードモデルを用いて、無作為化層別因子に加え、年齢(65歳未満、65歳以上)、人種(白人、白人以外)、ECOG PS(0、1)別のサブグループ解析を実施した。日本人集団は、全体集団と同様の解析手法により解析したが、層での調整については行わなかった。
多重性の調整:本試験では2回の中間解析及び最終解析を実施する計画とした。PFSは2回目の中間解析を最終解析とし、OSは2回の中間解析及び最終解析を実施することとした。PFS及びOSの有意水準は片側2.5%となるように厳密に制御した。これらの多重性の調整には、Maurer & Bretzのグラフィカルアプローチを用い、次のように計画した。まず、有意水準をPFSに片側2.5%配分する。PFSが統計学的に有意であった場合、有意水準をOSに再配分する。
2回の中間解析と最終解析における有意水準の配分には、α消費関数を用いることとした。
主な判定基準:
a: 治験担当医師がRECISTガイドライン1.1版に基づき評価、又は[RECISTガイドライン1.1版に基づく画像上の疾患進行(PD)が確認されていない場合]病理組織学的に確認したPDに基づく
b:治験担当医師がRECISTガイドライン1.1版に基づき評価
*1 未治療:子宮頸癌に対する根治的手術、放射線治療又は全身療法を受けておらず、かつ免疫療法による治療歴のない患者
*2 ⅠB2~ⅡB期:病巣が4cmを超えるもの、又は臨床的に明らかな病巣が子宮頸部を超えて広がっているが骨盤壁又は腟壁下1/3には達していないもの
*3 Ⅲ~ⅣA期: 腟壁浸潤が腟壁下にあるが骨盤側壁にまでは達していないもの、骨盤側壁にまで達しているもの若しくは水腎症や無機能腎を認めるもの、又は隣接する骨盤内臓器に浸潤があるもの
*4 FIGO 2018進行期分類のⅢ~ⅣA期に該当
FIGO 2014進行期分類からFIGO 2018進行期分類への読み替えについては表1、選択基準を満たすリンパ節転移陽性の定義は表2参照
*5 放射線治療:外部照射と小線源治療を合わせた全体の治療期間は50日(予期しない遅延のために最大56日間まで延長可能)を超えないこととした。
*6 ITT(intention to treat):無作為化されたすべての患者
*7 APaT(All Participants as Treated):無作為化され、治療を1回以上受けたすべての患者
*8 無作為化層別因子:計画された外部照射の種類(IMRT又はVMAT、IMRT又はVMAT以外)、スクリーニング時の病期[FIGO 2014進行期分類でⅠB2~ⅡB期(リンパ節転移陽性)、FIGO 2014進行期分類でⅢ~ⅣA期(リンパ節転移陽性又は陰性)]、計画された外部照射及び小線源治療の総線量[<70 Gy(2Gy換算等価線量(EQD2))、≧70 Gy(EQD2)]
表1 対象患者のFIGO 2018進行期分類
FIGO進行期分類は、2018年版において、リンパ節転移の有無が反映された分類となりました。骨盤リンパ節転移は、FIGO 2018進行期分類では新設されたⅢC1期、傍大動脈リンパ節転移は、FIGO 2018進行期分類では新設されたⅢC2期となりました1)。本試験の対象患者は、FIGO 2018進行期分類に基づくと、Ⅲ~ⅣA期に該当しました。

1)Bhatla N et al. Int J GynaecolObstet 2019; 145: 129-135
2)承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-A18試験)
3)日本産科婦人科学会・日本病理学会 編. 子宮頸癌取扱い規約 病理編 第5版, 2022年
表2 選択基準(抜粋)
以下のa又はbを満たす局所進行子宮頸癌患者

患者背景(ITT集団)

* CPS(combined positive score):PD-L1発現陽性細胞数(腫瘍細胞、リンパ球及びマクロファージ)を総腫瘍細胞数で除し、100を乗じた数値
(データカットオフ日:2023年1月9日)
主要評価項目:無増悪生存期間(PFS)(検証的解析結果)
副次評価項目:2年PFS率
■全体集団における無増悪生存期間:PFS(検証的解析結果)
■全体集団における2年PFS率



PFSのKaplan-Meier曲線(ITT集団)

NE:Not Estimated
治験担当医師がRECISTガイドライン1.1版に基づき評価、又は(RECISTガイドライン1.1版に基づく画像上のPDが確認されていない場合)病理組織学的に確認したPDに基づく
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化層別因子を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化層別因子を層別因子とした層別ログランク検定[片側]、有意水準α=0.0172(検証的解析結果)
無作為化層別因子:計画された外部照射の種類(IMRT又はVMAT、IMRT又はVMAT以外)、スクリーニング時の病期[FIGO 2014進行期分類でⅠB2~ⅡB期(リンパ節転移陽性)、FIGO 2014進行期分類でⅢ~ⅣA期(リンパ節転移陽性又は陰性)]、計画された外部照射及び小線源治療の総線量[<70 Gy(EQD2)、≧70 Gy(EQD2)]
(追跡期間中央値:キイトルーダ®+CCRT群 17.0ヵ月、プラセボ+CCRT群 17.0ヵ月、データカットオフ日:2023年1月9日)
主要評価項目:全生存期間(OS)(検証的解析結果)
副次評価項目:3年OS率
■全体集団における全生存期間:OS(検証的解析結果)
■全体集団における3年OS率

OSのKaplan-Meier曲線(ITT集団)

NE:Not Estimated
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化層別因子を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化層別因子を層別因子とした層別ログランク検定[片側]、有意水準α=0.01026(検証的解析結果)
無作為化層別因子:計画された外部照射の種類(IMRT又はVMAT、IMRT又はVMAT以外)、スクリーニング時の病期[FIGO 2014進行期分類でⅠB2~ⅡB期(リンパ節転移陽性)、FIGO 2014進行期分類でⅢ~ⅣA期(リンパ節転移陽性又は陰性)] 、計画された外部照射及び小線源治療の総線量[<70 Gy(EQD2)、≧70 Gy(EQD2)]
(追跡期間中央値:キイトルーダ®+CCRT群 27.8ヵ月、プラセボ+CCRT群 26.9ヵ月、データカットオフ日:2024年1月8日)
副次評価項目:CCRT実施後12週時点でのCR率/ORR
■CCRT実施後12週時点の完全奏効(CR)率
CR率(ITT集団のうち、組入れ時に測定可能病変を有する患者集団)

治験担当医師がRECISTガイドライン1.1版に基づき評価
(追跡期間中央値:キイトルーダ®+CCRT群 17.0ヵ月、プラセボ+CCRT群 17.0ヵ月、データカットオフ日:2023年1月9日)
■奏効率:ORR
ORR(ITT集団のうち、組入れ時に測定可能病変を有する患者集団)

*1 二項分布の確率計算による正確法
*2 層の例数を重みとし、無作為化層別因子を層別因子とした層別Miettinen and Nurminen法
*3 ベースライン後に画像評価を1回以上受けたがRECISTガイドライン1.1版に基づいた評価がなかった患者
*4 ベースライン後の画像評価がなかった患者
無作為化層別因子:計画された外部照射の種類(IMRT又はVMAT、IMRT又はVMAT以外)、スクリーニング時の病期[FIGO 2014進行期分類でⅠB2~ⅡB期(リンパ節転移陽性)、FIGO 2014進行期分類でⅢ~ⅣA期(リンパ節転移陽性又は陰性)] 、計画された外部照射及び小線源治療の総線量[<70 Gy(EQD2)、≧70 Gy(EQD2)]
治験担当医師がRECISTガイドライン1.1版に基づき評価
(追跡期間中央値:キイトルーダ®+CCRT群 17.0ヵ月、プラセボ+CCRT群 17.0ヵ月、データカットオフ日:2023年1月9日)
探索的評価項目:奏効期間(DOR)
■奏効期間:DOR

DORのKaplan-Meier曲線(ITT集団のうち、組入れ時に測定可能病変を有する患者集団)

Reprinted from The Lancet, Volume 403, Issue 10434, Lorusso D et al, Pembrolizumab or placebo with chemoradiotherapy followed by pembrolizumab or placebo for newly diagnosed, high-risk, locally advanced cervical cancer( ENGOT-cx11/GOG-3047/KEYNOTE-A18): a randomised, double-blind, phase 3 clinical trial, 1341-1350 Supplementary appendix 1, Copyright 2024, with permission from Elsevier.
治験担当医師がRECISTガイドライン1.1版に基づき評価

(追跡期間中央値:キイトルーダ®+CCRT群 17.0ヵ月、プラセボ+CCRT群 17.0ヵ月、データカットオフ日:2023年1月9日)
掲載誌の規定により、図の縦横比は原著通りとし、薬剤名(キイトルーダ®)は一般名(ペムブロリズマブ)で記載しています。
サブグループ解析:部分集団因子別の無増悪生存期間(PFS)
■部分集団因子別の無増悪生存期間:PFS


治験担当医師がRECISTガイドライン1.1版に基づき評価、又は(RECISTガイドライン1.1版に基づく画像上のPDが確認されていない場合)病理組織学的に確認したPDに基づく
(追跡期間中央値:キイトルーダ®+CCRT群 17.0ヵ月、プラセボ+CCRT群 17.0ヵ月、データカットオフ日:2023年1月9日)
サブグループ解析:部分集団因子別の全生存期間(OS)
■部分集団因子別の全生存期間:OS

(追跡期間中央値:キイトルーダ®+CCRT群 27.8ヵ月、プラセボ+CCRT群 26.9ヵ月、データカットオフ日:2024年1月8日)
注)第2回中間解析時のデータレビューに基づき、患者背景の一部が修正された
* 全体:投与群を共変量とし、無作為化層別因子を層別因子とした層別Cox比例ハザードモデルに基づく、サブグループ:投与群を共変量としたCox比例ハザードモデルに基づく
無作為化層別因子:計画された外部照射の種類(IMRT又はVMAT、IMRT又はVMAT以外)、スクリーニング時の病期[FIGO 2014進行期分類でⅠB2~ⅡB期(リンパ節転移陽性)、FIGO 2014進行期分類でⅢ~ⅣA期(リンパ節転移陽性又は陰性)] 、計画された外部照射及び小線源治療の総線量[<70 Gy(EQD2)、≧70 Gy(EQD2)]
全体集団 安全性(第1回中間解析)(APaT集団)
キイトルーダ®+CCRT群の副作用は507/528例(96.0%)に認められました。主な副作用(発現率20%以上)は、貧血313例(59.3%)、悪心302例(57.2%)、下痢266例(50.4%)、白血球数減少172例(32.6%)、好中球数減少153例(29.0%)、嘔吐132例(25.0%)、白血球減少症125例(23.7%)、血小板数減少116例(22.0%)、好中球減少症113例(21.4%)でした。重篤な副作用は91例(17.2%)に認められ、1%以上に認められた重篤な副作用は貧血14例(2.7%)、発熱9例(1.7%)、下痢8例(1.5%)でした。いずれかの治療の中止に至った副作用は81例(15.3%)に認められ、2例以上にみられたいずれかの治療の中止に至った副作用は、好中球減少症9例(1.7%)、白血球減少症、好中球数減少、白血球数減少 各7例(1.3%)、下痢6例(1.1%)、貧血5例(0.9%)、血小板減少症、アスパラギン酸アミノトランスフェラーゼ増加、血中クレアチニン増加、血小板数減少 各4例(0.8%)、肺臓炎、アラニンアミノトランスフェラーゼ増加 各3例(0.6%)、大腸炎2例(0.4%)でした。死亡に至った副作用は2例(0.4%)に認められ、内訳は、免疫性胃炎、大腸穿孔 各1例でした。
プラセボ+CCRT群では、副作用は509/530例(96.0%)に認められました。主な副作用(発現率20%以上)は、悪心315例(59.4%)、貧血292例(55.1%)、下痢271例(51.1%)、白血球数減少181例(34.2%)、嘔吐150例(28.3%)、好中球数減少148例(27.9%)、血小板数減少108例(20.4%)でした。重篤な副作用は65例(12.3%)に認められ、1%以上に認められた重篤な副作用は貧血7例(1.3%)でした。いずれかの治療の中止に至った副作用は67例(12.6%)に認められ、2例以上にみられたいずれかの治療の中止に至った副作用は、血中クレアチニン増加10例(1.9%)、好中球減少症6例(1.1%)、貧血、白血球減少症、好中球数減少、血小板数減少 各5例(0.9%)、悪心、クレアチニン腎クリアランス減少 各4例(0.8%)、血小板減少症、下痢、嘔吐、アラニンアミノトランスフェラーゼ増加、白血球数減少 各3例(0.6%)、リンパ球数減少、急性腎障害、腎機能不全 各2例(0.4%)でした。死亡に至った副作用は2例(0.4%)に認められ、内訳は、骨髄機能不全、好中球減少性大腸炎 各1例でした。
■主な副作用(いずれかの投与群で発現率5%以上)

MedDRA/J v25.1、GradeはCTCAE v5.0(データカットオフ日:2023年1月9日)
全体集団 安全性(第2回中間解析)(APaT集団)
キイトルーダ®+CCRT群の副作用は512/528例(97.0%)に認められました。主な副作用(発現率20%以上)は、貧血317例(60.0%)、悪心304例(57.6%)、下痢268例(50.8%)、白血球数減少173例(32.8%)、 好中球数減少156例(29.5%)、嘔吐135例(25.6%)、白血球減少症125例(23.7%)、血小板数減少116例(22.0%)、好中球減少症114例(21.6%)、甲状腺機能低下症112例(21.2%)でした。重篤な副作用は102例(19.3%)に認められ、1%以上に認められた重篤な副作用は貧血15例(2.8%)、発熱、下痢 各9例(1.7%)でした。いずれかの治療の中止に至った副作用は99例(18.8%)に認められ、2例以上にみられたいずれかの治療の中止に至った副作用は、貧血、好中球減少症 各9例(1.7%)、好中球数減少8例(1.5%)、白血球減少症、白血球数減少 各7例(1.3%)、血小板減少症、下痢 各6例(1.1%)、アスパラギン酸アミノトランスフェラーゼ増加、血中クレアチニン増加、血小板数減少、肺臓炎 各4例(0.8%)、アラニンアミノトランスフェラーゼ増加3例(0.6%)、大腸炎、免疫性胃炎 各2例(0.4%)でした。死亡に至った副作用は2例(0.4%)に認められ、内訳は、免疫性胃炎、大腸穿孔 各1例でした。
プラセボ+CCRT群では、副作用は513/530例(96.8%)に認められました。主な副作用(発現率20%以上)は、悪心317例(59.8%)、貧血296例(55.8%)、下痢271例(51.1%)、白血球数減少183例(34.5%)、嘔吐150例(28.3%)、好中球数減少148例(27.9%)、血小板数減少109例(20.6%)でした。重篤な副作用は71例(13.4%)に認められ、1%以上に認められた重篤な副作用は貧血7例(1.3%)でした。いずれかの治療の中止に至った副作用は69例(13.0%)に認められ、2例以上にみられたいずれかの治療の中止に至った副作用は、血中クレアチニン増加10例(1.9%)、貧血、好中球減少症 各6例(1.1%)、白血球減少症、血小板数減少 各5例(0.9%)、悪心、クレアチニン腎クリアランス減少、好中球数減少 各4例(0.8%)、血小板減少症、下痢、嘔吐、アラニンアミノトランスフェラーゼ増加、白血球数減少 各3例(0.6%)、リンパ球数減少、急性腎障害、腎機能不全 各2例(0.4%)でした。死亡に至った副作用は2例(0.4%)に認められ、内訳は、骨髄機能不全、好中球減少性大腸炎 各1例でした。
■主な副作用(いずれかの投与群で発現率5%以上)

MedDRA/J v26.1、GradeはCTCAE v5.0(データカットオフ日:2024年1月8日)
■免疫関連など特に注目すべき有害事象(APaT集団)
(いずれかの投与群で発現)

MedDRA/J v25.1、GradeはCTCAE v5.0(データカットオフ日:2023年1月9日)
■免疫関連など特に注目すべき有害事象(APaT集団)
(いずれかの投与群で発現)

MedDRA/J v26.1、GradeはCTCAE v5.0(データカットオフ日:2024年1月8日)
関連コンテンツ
キイトルーダ®・悪性腫瘍関連領域情報


キイトルーダ®治療日誌:<頭頸部癌>キイトルーダ®術前補助療法
