KEYNOTE-177試験
MSI-High結腸・直腸癌:国際共同臨床試験成績:国際共同第Ⅲ相試験〈KEYNOTE-177試験〉
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
André T et al. N Engl J Med 2020; 383: 2207-2218
本試験はMSD社の資金提供により行われた。Thierry AndréはMSD社から顧問料などを受領している。
また、著者のうち、Ping Yang、 Mohammed Z H Farooqui、Patricia Marinello、Luis A Diaz Jrは同社の社員である。その他の著者にMSD社より講演料、顧問料などを受領している者が含まれる。
Andre T et al. Lancet Oncol 2021; 22: 665-677
本試験はMSD社の資金提供により行われた。Thierry AndreはMSD社から顧問料などを受領している。
また、著者のうち、Mayur Amonkar、Josephine M Norquist、Ping Yang、Mohammed Farooquiは同社の社員である。その他の著者にMSD社より講演料、顧問料などを受領している者が含まれる。
試験概要
【目的】未治療のMMR欠損又はMSI-Highの結腸・直腸癌(Ⅳ期)の一次治療として、キイトルーダ®による有効性及び安全性を標準療法と比較検討する。
【デザイン】国際共同無作為化非盲検第Ⅲ相試験[優越性検証試験]
【対象】未治療のMMR欠損又はMSI-High*の結腸・直腸癌(Ⅳ期)患者307例(キイトルーダ®群153例、標準療法群(対照群)154例;日本人患者22例[キイトルーダ®群12例、標準療法群(対照群)10例]を含む)
【方法】対象患者をキイトルーダ®群又は標準療法群(対照群)のいずれかに1:1の割合で無作為に割り付けた。キイトルーダ®群では、キイトルーダ®200mgを3週間間隔(Q3W)で30分かけて静脈内投与し、最大35サイクル(約2年間)まで継続した。標準療法群(対照群)では、治験担当医師の選択によりmFOLFOX6、mFOLFOX6+ベバシズマブ、mFOLFOX6+セツキシマブ、FOLFIRI、FOLFIRI+ベバシズマブ又はFOLFIRI+セツキシマブのいずれかを2週間間隔(Q2W)で静脈内投与し、独立中央画像判定機関がRECIST1.1に基づき疾患進行(PD)と判定し、クロスオーバーに関する基準#をすべて満たした場合は、任意でキイトルーダ®の単独投与を最大35サイクルまで受けることを可能とした。
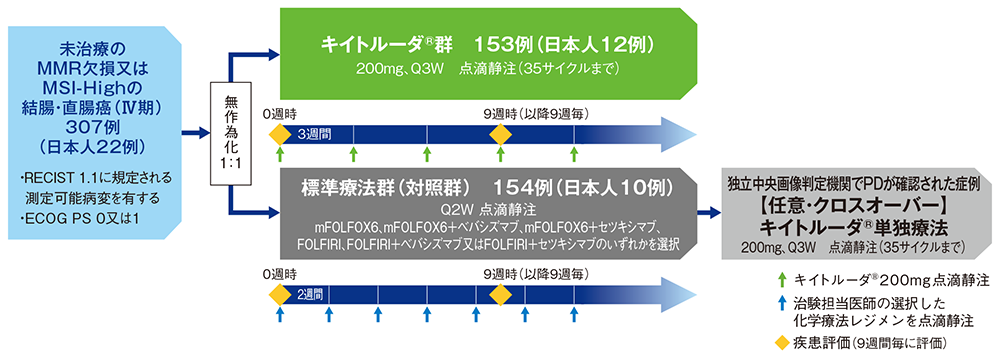
- 無作為化にあたって、年齢、性別、その他の患者特性による層別化は行わなかった。
- 画像評価は無作為化に先立つ28日以内、無作為化後は、9週毎に行い、臨床的に必要とされた場合はより早い時期に実施した。
- キイトルーダ®投与第2期として以下の場合に追加のキイトルーダ®投与が認められた。
【キイトルーダ®群】
実施医療機関によりCRが確定し投与を中止した患者又は最大35回の投与を完了し、かつ、SD以上を示した患者において、キイトルーダ®の投与終了後にPDが確認された場合、投与第2期で更に17回の投与を許容した。
【標準療法群(対照群)】
クロスオーバー期に移行し、キイトルーダ®の投与終了後(最大35回)にPDが確認された場合、事前に規定した基準を満たしていれば、投与第2期で更に17回の投与を許容した。
# クロスオーバーに関する主な基準
・RECIST 1.1により盲検下の独立中央画像判定機関がPDと判定 ・治験薬の最終投与以降、がんに対する他の治療を受けていない ・化学療法レジメン及びベバシズマブ/セツキシマブによる有害事象がGrade 1(CTCAE ver4.0)以下に回復している ・ECOG PS 0又は1 ・適切な臓器機能を有する ・妊娠の可能性のある女性患者の場合、キイトルーダ®の投与開始前72時間以内の血清妊娠検査が陰性
【主要評価項目】無増悪生存期間(PFS)※、全生存期間(OS)※
※検証的解析項目
【副次評価項目】奏効率(ORR)、安全性
【探索的評価項目】奏効期間(DOR)、無作為化から二次治療開始後のPD又は死亡のいずれか早い時点までの期間(PFS2)、健康関連QOLなど
【判定基準】PFS、ORR及びDORは、独立中央画像判定機関がRECISTガイドライン1.1版に基づき判定した。
健康関連QOLに関するPROは、EORTC QLQ-C30、EORTC QLQ-CR29及びEQ-5Dの質問票を用いて評価した。
【解析計画】解析対象集団:有効性の解析はITT集団(無作為化されたすべての患者)、健康関連QOLの解析はFAS集団(1回以上治験薬を投与され、かつ、1回以上PROの評価を実施した患者)、安全性の解析はASaT集団(1回以上治験薬を投与されたすべての患者)を対象として実施した。
有効性評価の統計手法:有効性の主要評価項目であるPFS及びOSのいずれかでキイトルーダ®群が標準療法群(対照群)に対して優越性を示した場合に主要目的を達成したとみなした。PFS及びOSはKaplan-Meier法を用いて各投与群の生存曲線を推定した。群間差はログランク検定を用いて評価しp値を算出した。投与群を共変量としてモデルに含めたCox比例ハザードモデルに基づきハザード比及びその95%CIを算出した。PFSは最終解析であり、OSは中間解析である。約190件のOSのイベントが観察された時点、又は2回目の中間解析実施後12ヵ月時点のいずれか早い時点でOSの最終解析が行われる。副次評価項目のORRは、OS又はPFSの帰無仮説が棄却された場合に検定することとし、群間比較にはMiettinen and Nurminen法を用いた。両群間のORRの差の95%CIを算出した。探索的評価項目であるDORはKaplan-Meier法を用いて推定し、PFS2は無作為化から二次治療開始後のPD又は死亡のいずれか早い時点までの期間と定義し解析した。サブグループ解析として、事前規定された年齢、性別、ECOG PS、地域、転移(肝/肺転移又はその他の転移)、診断(再発又は新規)、BRAF遺伝子変異(野生型又はBRAF V600E遺伝子変異)、KRAS/NRAS遺伝子変異(野生型又はKRAS/NRAS遺伝子変異)、原発巣(右又は左)を対象としてCox比例ハザードモデルに基づきPFS、OSの部分集団解析を実施した。日本人集団におけるPFS、OS、ORR、DORは、治験実施計画書には記載されていないが、全体集団と同様の統計手法にて検討し、評価資料として承認時に評価された。
健康関連QOLに関するPRO:QLQ-C30及びEQ-5Dのベースラインからの変化量(18週時)を解析した。また、EORTC QLQ-CR29の機能スコア及び症状スコアのベースライン時からの変化を解析した。
【データカットオフ】2020年2月19日(2回目の中間解析)
*MMR欠損又はMSI-Highの検査は、無作為化割付け42~1日前のスクリーニング期間に行った。
ホリナートの効能又は効果、用法及び用量は以下のとおりです。
効能又は効果 ホリナート・テガフール・ウラシル療法
結腸・直腸癌に対するテガフール・ウラシルの抗腫瘍効果の増強
用法及び用量 ホリナート・テガフール・ウラシル療法
通常、成人にはホリナートとして75mgを、1日3回に分けて(約8時間ごとに)、テガフール・ウラシル配合剤と同時に経口投与する。
テガフール・ウラシル配合剤の投与量は、通常、1日量として、テガフール300〜600mg相当量(300mg/m2を基準)を1日3回に分けて(約8時間ごとに)、食事の前後1時間を避けて経口投与する。以上を28日間連日経口投与し、その後7日間休薬する。
これを1クールとして投与を繰り返す。
レボホリナートの効能又は効果は以下のとおりです。
効能又は効果 レボホリナート・フルオロウラシル療法
胃癌(手術不能又は再発)及び結腸・直腸癌に対するフルオロウラシルの抗腫瘍効果の増強
レボホリナート・フルオロウラシル持続静注併用療法
結腸・直腸癌、小腸癌及び治癒切除不能な膵癌に対するフルオロウラシルの抗腫瘍効果の増強
フルオロウラシル(5-FU)の効能又は効果、用法及び用量は以下のとおりです。
4. 効能又は効果(抜粋)
○レボホリナート・フルオロウラシル持続静注併用療法
結腸・直腸癌、小腸癌、治癒切除不能な膵癌
6. 用法及び用量(抜粋)
6.4 結腸・直腸癌に対するレボホリナート・フルオロウラシル持続静注併用療法
3)通常、成人にはレボホリナートとして1回200mg/m2(体表面積)を2時間かけて点滴静脈内注射する。レボホリナートの点滴静脈内注射終了直後にフルオロウラシルとして400mg/m2(体表面積)を静脈内注射、さらにフルオロウラシルとして2400〜3000mg/m2(体表面積)を46時間持続静注する。これを2週間ごとに繰り返す。
なお、年齢、患者の状態などにより適宜減量する。
セツキシマブの効能又は効果は以下のとおりです。
効能又は効果(抜粋)
RAS遺伝子野生型の治癒切除不能な進行・再発の結腸・直腸癌
イリノテカンの用法及び用量は以下のとおりです。
用法及び用量(抜粋)
結腸・直腸癌(手術不能又は再発)はA法又はB法を使用する。
A法 イリノテカン塩酸塩水和物として、通常、成人に1日1回、100mg/m2を1週間間隔で3〜4回点滴静注し、少なくとも2週間休薬する。これを1クールとして、投与を繰り返す。
B法 イリノテカン塩酸塩水和物として、通常、成人に1日1回、150mg/m2を2週間間隔で2〜3回点滴静注し、少なくとも3週間休薬する。これを1クールとして、投与を繰り返す。
4. 効能又は効果(抜粋)
治癒切除不能な進行・再発の高頻度マイクロサテライト不安定性(MSI-High)を有する結腸・直腸癌
5. 効能又は効果に関連する注意(抜粋)
〈治癒切除不能な進行・再発のMSI-Highを有する結腸・直腸癌〉
5.19 十分な経験を有する病理医又は検査施設における検査により、MSI-Highが確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること。なお、承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
5.20 本剤の術後補助療法における有効性及び安全性は確立していない。
患者背景(ITT集団)
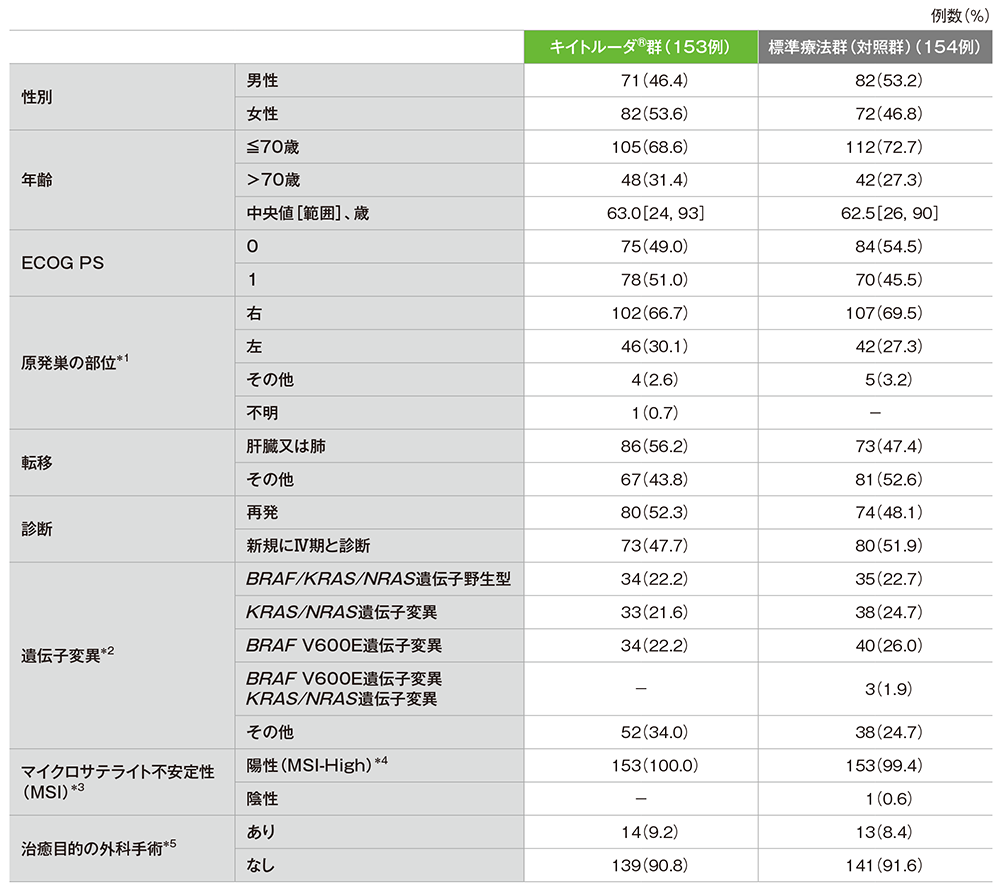
*1 両側に原発巣が見られる場合は「その他」とした。
*2
BRAF V600E、KRAS、NRAS遺伝子のうち測定できた遺伝子でいずれにも変異が認められなかった場合で、変異ステータスが不明(未確定又は欠損)の遺伝子が1つ以上あるか、もしくはBRAF遺伝子変異のタイプがV600Eでない場合は、「その他」に分類した。
*3 MSIは各施設の検査室でPCR検査又はIHC検査を用いて判定した。
*4 陽性(MSI-High)の定義は以下の通りとする。
• MSI検査において、MSIマーカー3〜5つのうち、2つ以上のマーカーがMSI+を示した場合
• IHC検査において、MMRタンパク質(MLH1、MSH2、MSH6、PMS2)の発現が1つ以上欠損している場合
*5 無作為化から、新たな抗がん剤療法、クロスオーバー治療及びキイトルーダ®投与第2期までに行った治癒目的の手術。
●標準療法群(対照群)における免疫チェックポイント阻害剤の投与
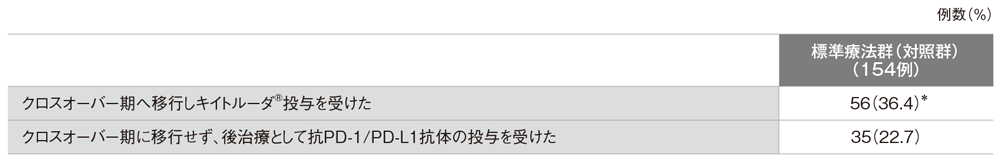
*データカットオフ時にキイトルーダ®の投与を継続していた患者は17例(30.4%)であった。
主要評価項目(優越性試験):無増悪生存期間(PFS)(ITT集団)
●PFSのKaplan-Meier曲線
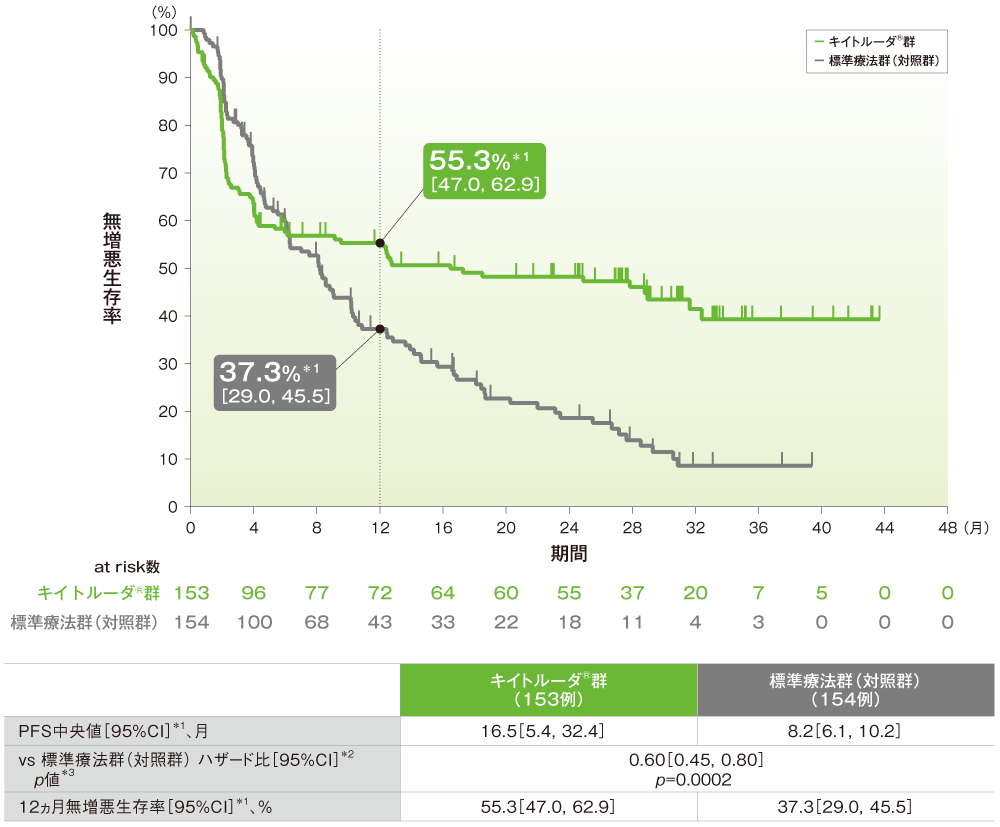
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、Efron法を用いたCox回帰モデルに基づく
*3 ログランク検定[片側]、有意水準α=0.0117
データカットオフ:2020年2月19日(2回目の中間解析)
(追跡期間中央値:キイトルーダ®群28.4ヵ月(範囲:0.2, 48.3)、標準療法群(対照群)27.2ヵ月(範囲:0.8, 46.6))
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
André T et al. N Engl J Med 2020; 383: 2207-2218
本試験はMSD社の資金提供により行われた。著者にMSD社社員、MSD社より講演料、顧問料などを受領している者が含まれる。
- PFS中央値はキイトルーダ®群が16.5ヵ月(95%CI:5.4, 32.4)、標準療法群(対照群)が8.2ヵ月(95%CI:6.1, 10.2)であり、有意にPFSを改善しました(p=0.0002、ログランク検定[片側]、有意水準α=0.0117;検証的解析結果)。
- 12ヵ月無増悪生存率はキイトルーダ®群が55.3%(95%CI: 47.0, 62.9)、標準療法群(対照群)が37.3%(95%CI: 29.0, 45.5)でした。
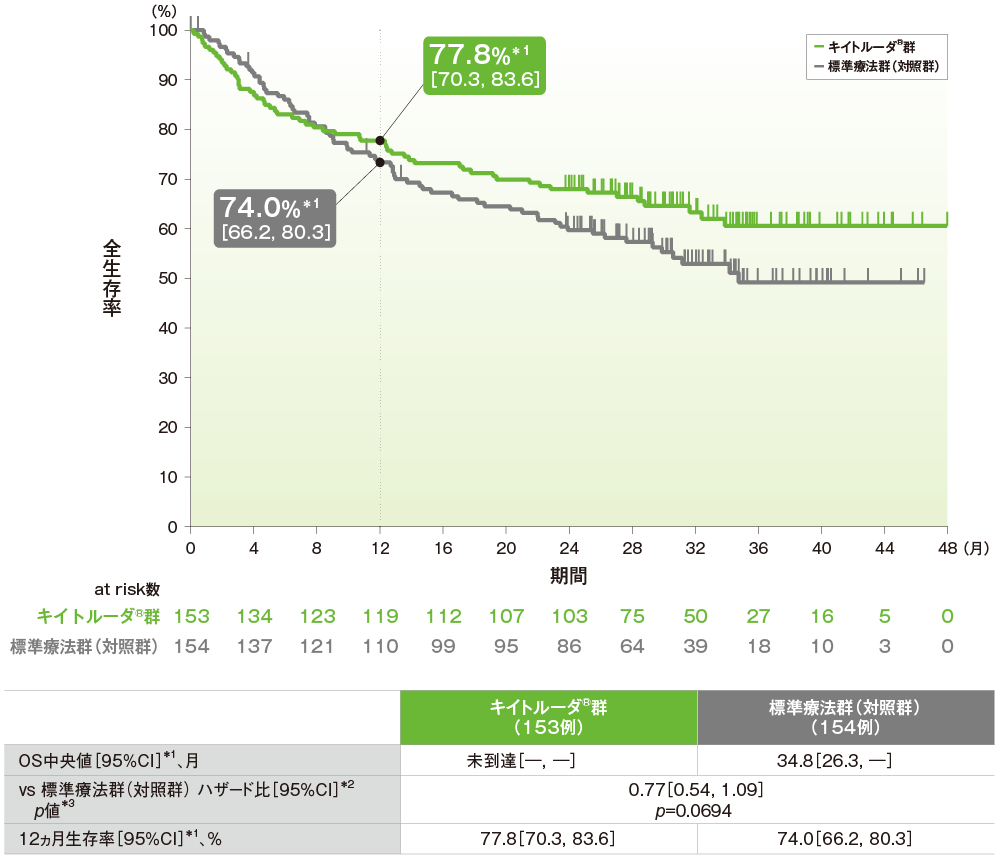
主要評価項目(優越性試験):全生存期間(OS)(ITT集団)
全生存期間(OS)は中間解析結果を示す(データカットオフ日: 2020年2月19日[2回目の中間解析])
●OSのKaplan-Meier曲線
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、Efron法を用いたCox回帰モデルに基づく
*3 ログランク検定[片側]、有意水準α=0.0053
データカットオフ:2020年2月19日(2回目の中間解析)
(追跡期間中央値:キイトルーダ®群28.4ヵ月(範囲:0.2, 48.3)、標準療法群(対照群)27.2ヵ月(範囲:0.8, 46.6))
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
André T et al. N Engl J Med 2020; 383: 2207-2218
本試験はMSD社の資金提供により行われた。著者にMSD社社員、MSD社より講演料、顧問料などを受領している者が含まれる。
- OS中央値はキイトルーダ®群が未到達(95%CI:未到達, 未到達)、標準療法群(対照群)が34.8ヵ月(95%CI:26.3, 未到達)であり、優越性は検証されませんでした(検証的解析結果)。
- 12ヵ月生存率はキイトルーダ®群が77.8%(95%CI:70.3, 83.6)、標準療法群(対照群)が74.0%(95%CI:66.2, 80.3)でした。
探索的評価項目:無作為化から二次治療開始後のPD又は死亡のいずれか早い時点までの期間(PFS2)(ITT集団)
- PFS2中央値は、キイトルーダ®群で未到達、標準療法群(対照群)で23.5ヵ月(95%CI:16.6, 32.6)、標準療法群(対照群)に対するキイトルーダ®群のPFS2のハザード比は0.63(95%CI:0.45, 0.88)でした。
データカットオフ:2020年2月19日(2回目の中間解析)
(追跡期間中央値:キイトルーダ®群28.4ヵ月(範囲:0.2, 48.3)、標準療法群(対照群)27.2ヵ月(範囲:0.8, 46.6))
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
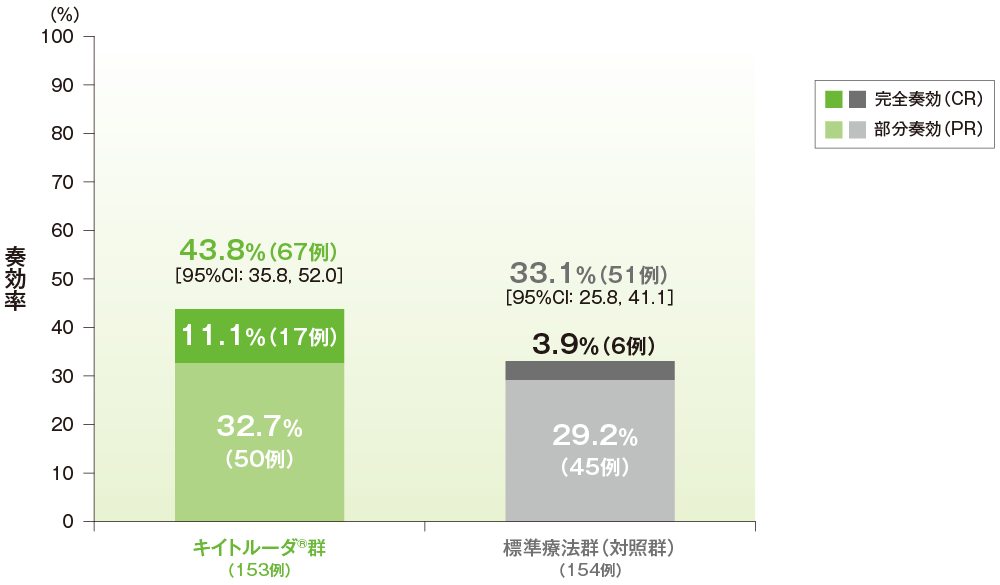
副次評価項目:奏効率(ORR)(ITT集団)
●奏効率(ORR)
●奏効率(ORR)の要約
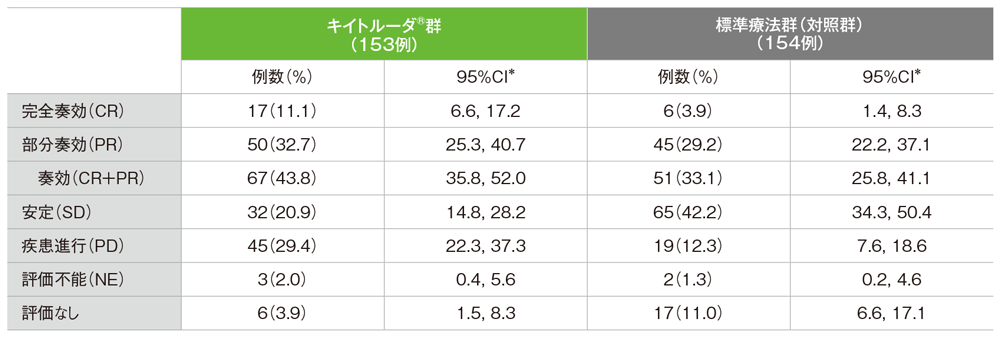
独立中央画像判定機関がRECISTガイドライン1.1版に基づき判定
*二項分布の確率計算による正確法
データカットオフ:2020年2月19日(2回目の中間解析)
(追跡期間中央値:キイトルーダ®群28.4ヵ月(範囲:0.2, 48.3)、標準療法群(対照群)27.2ヵ月(範囲:0.8, 46.6))
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
André T et al. N Engl J Med 2020; 383: 2207-2218
本試験はMSD社の資金提供により行われた。著者にMSD社社員、MSD社より講演料、顧問料などを受領している者が含まれる。
- ORRはキイトルーダ®群が43.8%(95%CI:35.8, 52.0)、標準療法群(対照群)が33.1%(95%CI:25.8, 41.1)でした。
探索的評価項目:奏効期間(DOR)(ITT集団)
●奏効までの期間及び奏効期間(DOR)の要約
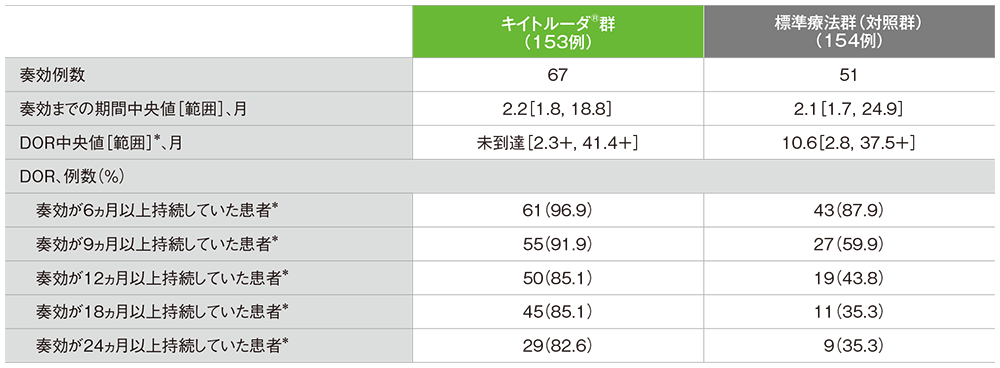
独立中央画像判定機関がRECISTガイドライン1.1版に基づき判定
*打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく。「+」は最後の疾患評価までにPDがみられないことを示す
データカットオフ:2020年2月19日(2回目の中間解析)
(追跡期間中央値:キイトルーダ®群28.4ヵ月(範囲:0.2, 48.3)、標準療法群(対照群)27.2ヵ月(範囲:0.8, 46.6))
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
- DOR中央値はキイトルーダ®群が未到達(範囲:2.3+, 41.4+)、標準療法群(対照群)が10.6ヵ月(範囲:2.8, 37.5+)でした。
- DORが24ヵ月以上であったのは、キイトルーダ®群が29例(82.6%)、標準療法群(対照群)が9例(35.3%)でした。
安全性(ASaT集団)
●副作用
キイトルーダ®群
- 副作用は122/153例(79.7%)に認められました。主な副作用(発現率10%以上)は、下痢38例(24.8%)、疲労32例(20.9%)、そう痒症21例(13.7%)、悪心19例(12.4%)、アスパラギン酸アミノトランスフェラーゼ増加、発疹各17例(11.1%)、関節痛、甲状腺機能低下症各16例(10.5%)でした。
重篤な副作用
- 重篤な副作用は25/153例(16.3%)に認められました。発現率1%以上の重篤な副作用は、大腸炎3例(2.0%)、急性腎障害、自己免疫性大腸炎、下痢、肝炎、発熱が各2例(1.3%)でした。
投与中止に至った副作用
- 投与中止に至った副作用は15/153例(9.8%)に認められました。内訳は、アラニンアミノトランスフェラーゼ増加、自己免疫性大腸炎、大腸炎及び肝炎各2例(1.3%)、自己免疫性肝炎、急性腎障害、免疫性肝炎、肺臓炎、乾癬、下垂体炎、アスパラギン酸アミノトランスフェラーゼ増加各1例(0.7%)でした。
死亡に至った副作用
- 本試験では、死亡に至った副作用は認められませんでした。
標準療法群(対照群)
- 副作用は141例/ 143例(98.6%)に認められました。主な副作用(発現率10%以上)は悪心79例(55.2%)、下痢75例(52.4%)、疲労63例(44.1%)、食欲減退49例(34.3%)、口内炎43例(30.1%)、嘔吐40例(28.0%)、好中球数減少33例(23.1%)、好中球減少症30例(21.0%)、末梢性感覚ニューロパチー29例(20.3%)、脱毛症28例(19.6%)、無力症、粘膜の炎症、末梢性ニューロパチー、手掌・足底発赤知覚不全症候群各25例(17.5%)、鼻出血20例(14.0%)、貧血、白血球数減少各17例(11.9%)、浮動性めまい15例(10.5%)でした。
重篤な副作用
- 重篤な副作用は、41/143例(28.7%)に認められました。発現率1%以上の重篤な副作用は、下痢9例(6.3%)、発熱性好中球減少症5例(3.5%)、食欲減退、疲労、好中球減少症各3例(2.1%)、急性腎障害、腸管穿孔、嘔吐が各2例(1.4%)でした。
投与中止に至った副作用
- 投与中止に至った副作用は8/143例(5.6%)に認められました。内訳は、急性心筋梗塞、下痢、脳血管発作、疲労、腸管穿孔、口内炎、発熱、無力症、発熱性好中球減少症各1例(0.7%)でした。
死亡に至った副作用
- 死亡に至った副作用は1/143例(0.7%)に認められました。内訳は、腸管穿孔1例(0.7%)でした。
●副作用(ASaT集団)(いずれかの群で発現率5%以上)
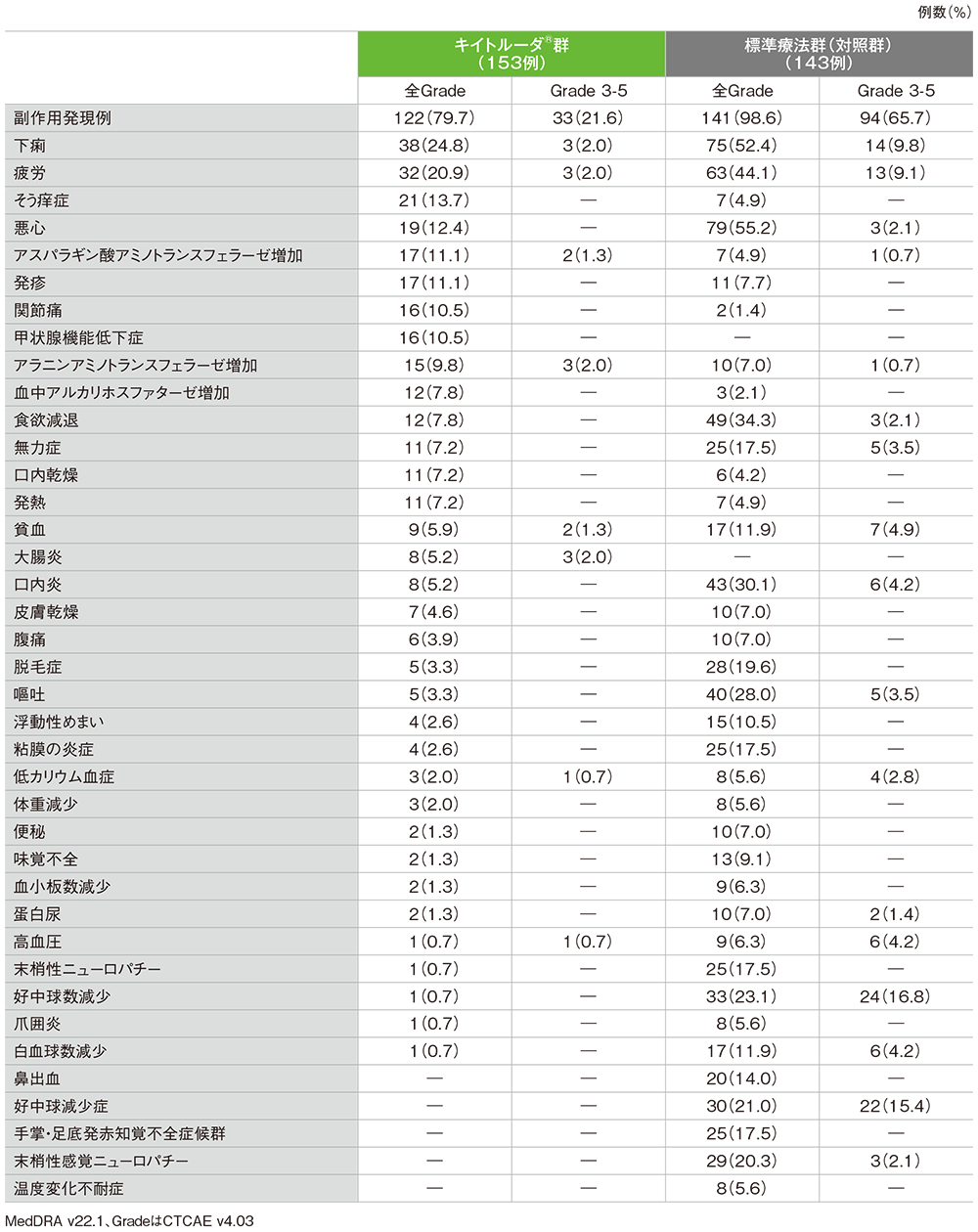
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-177試験)
キイトルーダ®・悪性腫瘍関連領域情報
関連製品
レンビマ®
