〈アキシチニブ併用〉国際共同第Ⅲ相試験<KEYNOTE-426試験>(全集団)
腎細胞癌:国際共同臨床試験成績:国際共同第Ⅲ相試験<KEYNOTE-426試験>
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
Rini BI et al. N Engl J Med 2019; 380: 1116-1127#1
Powles T et al. Lancet Oncol 2020; 21: 1563-1573#2
Plimack ER et al. Eur Urol 2023; 84: 449-454#3
#1 KEYNOTE-426試験はMSD社の資金提供により行われた。Brian I. RiniはMSD社から顧問料などを受領している。その他の著者に同社から顧問料などを受領している者が含まれる。本試験はアカデミックアドバイザーとMSD社の社員によりデザインされた。著者のうち、Qiong Shou、Rodolfo F. Perini、Mei ChenはMSD社の社員である。
#2 第2回中間解析において、Thomas PowlesはMSD社から研究費などを受領している。その他の著者に同社から研究費などを受領している者が含まれる。著者のうち、Lina Yin、Mei Chen、L Rhoda MolifeはMSD社の社員である。
#3 最終解析において、Elizabeth R. PlimackはMSD社から研究費などを受領している。その他の著者に同社から研究費などを受領している者が含まれる。Chenxiang Li、Rodolfo Perini、L. Rhoda MolifeはMSD社の社員である。
試験概要
【目的】化学療法未治療の局所進行性又は転移性腎細胞癌患者におけるキイトルーダ®+アキシチニブ※併用とスニチニブ単独の有効性及び安全性を比較検討する。
【デザイン】国際共同無作為化非盲検第Ⅲ相試験[優越性検証試験]
[第1回中間解析結果(データカットオフ日:2018年8月24日)、第2回中間解析結果(データカットオフ日:2020年1月6日)、最終解析結果(データカットオフ日:2021年1月11日)]
【対象】化学療法未治療の局所進行性又は転移性腎細胞癌(淡明細胞型)患者861例(日本人94例を含む)
【方法】キイトルーダ®+アキシチニブ※併用群[キイトルーダ®200mgを3週間間隔(Q3W)で点滴静注とアキシチニブ※5mgを1日2回(BID)で経口投与]又はスニチニブ群[50mgを1日1回(QD)で4週間経口投与後、2週間休薬]に1:1の割合で無作為に割り付けた。画像診断をベースライン時、12週時、その後6週間毎、54週以降は12週間毎に実施し、抗腫瘍効果を評価した。疾患進行、許容できない有害事象の発現、医師又は患者による中止決定まで投与を継続した(キイトルーダ®投与は最大35サイクル)。
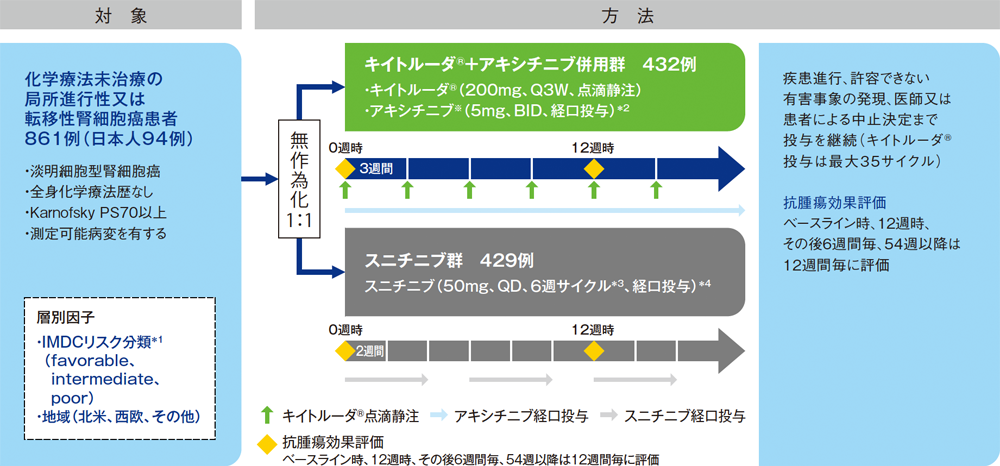
【評価項目】主要評価項目:全生存期間(overall survival;OS)※及び無増悪生存期間(progression free survival;PFS)※
副次評価項目:奏効率(overall response rate;ORR)、病勢コントロール率(disease control rate;DCR)、奏効期間(duration of response;DOR)、安全性 など
※第1回中間解析は検証的解析項目
【判定基準】PFS、ORRについて、RECISTガイドライン1.1版に基づき盲検下独立判定委員会(blinded independent central review;BICR)が評価した。
【解析計画】解析対象集団:有効性はITT集団*5、安全性はASaT集団*6を解析対象とした。
統計手法:OS、PFSの生存曲線はKaplan-Meier法を用いて推定した。OS、PFSの群間比較は層別ログランク検定、ハザード比は投与群のみを共変量とした層別Cox比例ハザードモデルにより評価した。層別Cox比例ハザードモデルを用いてOS及びPFSのカテゴリー[年齢(<65歳、≧65歳)、性別(男性、女性)、人種(白人、白人以外)、地域(北米、西欧、その他)、IMDCリスク分類(Favorable、Intermediate、Poor;Favorable、Intermediate又はPoor)、Karnofsky PS(90又は100、70又は80)、PD-L1発現(CPS<1、CPS≧1)、転移臓器数(1、≧2)]ごとの部分集団解析を実施した。ORRの群間比較は、層の例数で重み付けした層別Miettinen and Nurminen法により評価した。各国の規制要件に応じて、各国の集団を解析する可能性ありの規定に基づき、日本人集団については、OS、PFS、ORR、DCR、DORについて算出し、評価資料として承認時に評価された。
多重性の調整:本試験は2回の中間解析を事前に計画し、試験全体の有意水準を片側2.5%に厳密に制御した。1回目の中間解析では、OS及びPFSを解析し、いずれかの優越性が示された場合のみ、ORRを解析することとした。2回目の中間解析ではOS及びPFS、最終解析ではOSを解析することとした*7。2つの主要仮説の検定について、PFSはHwang-Shih-DeCani(γ=−2)のα消費関数を用いた群逐次検定、OSは1回目の中間解析の有意水準を0.01%に固定し、残りの有意水準はHwang-Shih-DeCani(γ=−4)のα消費関数を用いた群逐次検定を行った。PFS、OS、ORRについて、Maurer & BretzのGraphical approachにより多重性を調整した。
各評価項目は以下のとおり検定される可能性があった。
PFS:最初に有意水準0.2%を割り当てる。OSのみ有意となった場合、OSに割り当てられた有意水準の2.3%の半分を再配分して、1.35%で検定する。OS及びORRのいずれも有意となった場合、すべての有意水準を再配分し2.5%で検定する。
OS:最初に有意水準2.3%を割り当てる。PFS及びORRのいずれも有意となった場合、すべての有意水準を再配分し2.5%で検定する。
ORR:OS又はPFSのいずれかが有意になった場合のみ検定する。PFSのみ有意となった場合、PFSに割り当てられた有意水準を再配分して0.2%で検定する。OSのみ有意となった場合、OSに割り当てられた有意水準の2.3%の半分を再配分して、1.15%で検定する。OS及びPFSのいずれも有意となった場合、すべての有意水準を再配分し2.5%で検定する。
*1 IMDC(International Metastatic Renal Cell Carcinoma Database Consortium)リスク分類:6つのリスク因子[Karnofsky PS80未満、初診から治療開始までの期間(本試験の無作為化までの期間)が1年未満、ヘモグロビン値が正常範囲の下限未満、補正血清カルシウム値が正常範囲の上限超、好中球絶対数が正常範囲の上限超、血小板数が正常範囲の上限超]で構成され、IMDCスコア0をfavorable、1-2をintermediate、3-6をpoorに分類
*2 安全性基準[5mg BIDが連続する2コース(6週間)以上忍容であり、Grade 2を超えるアキシチニブの副作用が認められず、血圧が150/90mmHg以下に管理された場合]を満たす場合に7mg BID、同様の基準で10mg BIDへの増量、副作用管理を目的として3mg BID、2mg BIDへの減量を可能とした
*3 4週間連日投与後、2週間休薬
*4 副作用管理を目的として各6週サイクルの最初の4週間は37.5mg QD、25mg QDへの減量を可能とした
*5 ITT(intention to treat)集団:無作為化されたすべての患者
*6 ASaT(all subjects as treated)集団:治験薬が1回以上投与されたすべての患者
*7 1回目の中間解析でいずれの評価項目も優越性が示された場合、その後の検定は実施されない
4. 効能又は効果(抜粋)
根治切除不能又は転移性の腎細胞癌
※アキシチニブの効能又は効果、用法及び用量は以下のとおりです。
4. 効能又は効果
根治切除不能又は転移性の腎細胞癌
6. 用法及び用量
通常、成人にはアキシチニブとして1回5mgを1日2回経口投与する。なお、患者の状態により適宜増減するが、1回10mg1日2回まで増量できる。
7. 用法及び用量に関連する注意
7.1 抗悪性腫瘍剤(サイトカイン製剤を含む)による治療歴のない患者に対しては、PD-1/PD-L1阻害剤と併用すること。
7.2 1回5mg1日2回、2週間連続投与し、本剤に忍容性が認められる場合には、1回7mg1日2回投与に増量することができる。連続2週間投与して本剤に忍容性が認められる場合には、更に最大1回10mg1日2回に増量することができる。
7.3 副作用がみられた場合は、必要に応じて、本剤を減量、休薬又は中止すること。減量して投与を継続する場合は、副作用の症状、重症度等に応じて、1回3mg1日2回、又は1回2mg1日2回に減量すること。
患者背景(ITT集団)
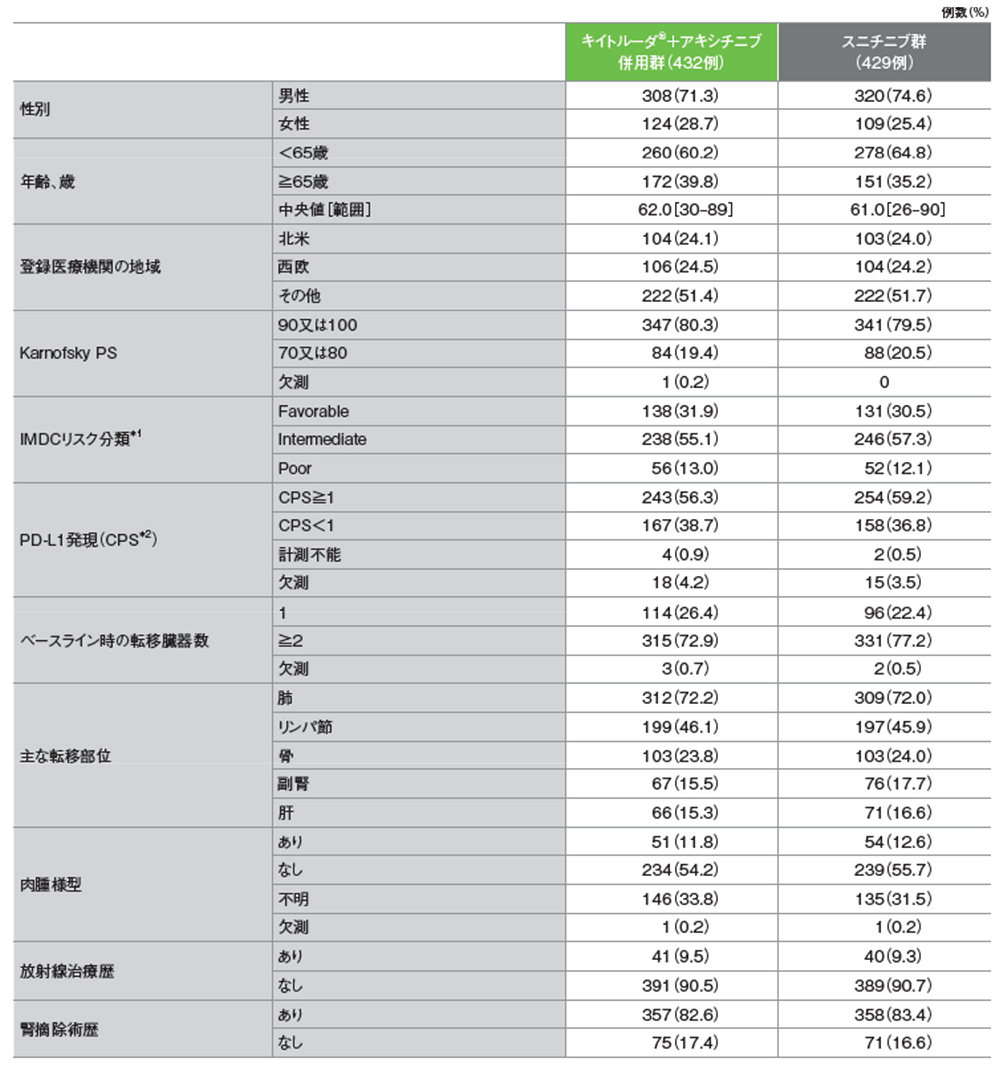
*1 IMDC(International Metastatic Renal Cell Carcinoma Database Consortium)リスク分類:6つのリスク因子[Karnofsky PS80未満、初診から治療開始までの期間(本試験の無作為化までの期間)が1年未満、ヘモグロビン値が正常範囲の下限未満、補正血清カルシウム値が正常範囲の上限超、好中球絶対数が正常範囲の上限超、血小板数が正常範囲の上限超]で構成され、IMDCスコア0をfavorable、1-2をintermediate、3-6をpoorに分類
*2 CPS(combined positive score):PD-L1陽性細胞数(腫瘍細胞、リンパ球及びマクロファージ)を総腫瘍細胞数で除し、100を乗じた数値
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
主要評価項目 全生存期間:OS
全生存期間(OS)のKaplan-Meier曲線(ITT集団;検証的解析結果)

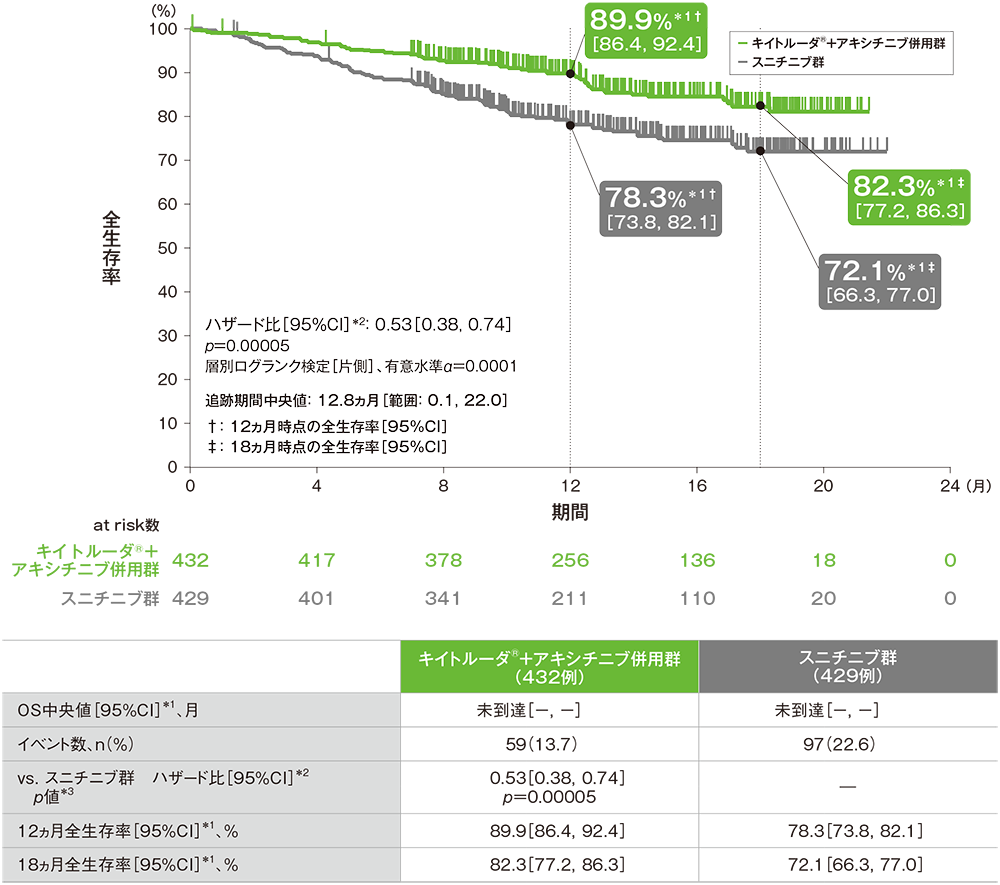
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子(無作為化に用いた層別因子)とした層別Cox比例ハザードモデルに基づく
*3 層別ログランク検定[片側](層別因子は無作為化に用いた層別因子)、有意水準α=0.0001
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
- 第1回中間解析において、キイトルーダ®+アキシチニブ併用群はスニチニブ群に対して、OSを有意に延長しました(ハザード比:0.53、95%CI:0.38, 0.74、p=0.00005、層別ログランク検定[片側]、有意水準α=0.0001;検証的解析結果)。
OS中央値はキイトルーダ®+アキシチニブ併用群及びスニチニブ群ともに未到達であり、12ヵ月時点の全生存率はそれぞれ89.9%(95%CI:86.4, 92.4)、78.3%(95%CI:73.8, 82.1)、18ヵ月時点の全生存率はそれぞれ82.3%(95%CI:77.2, 86.3)、72.1%(95%CI:66.3, 77.0)でした。
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
全⽣存期間(OS)のKaplan-Meier曲線(ITT集団)

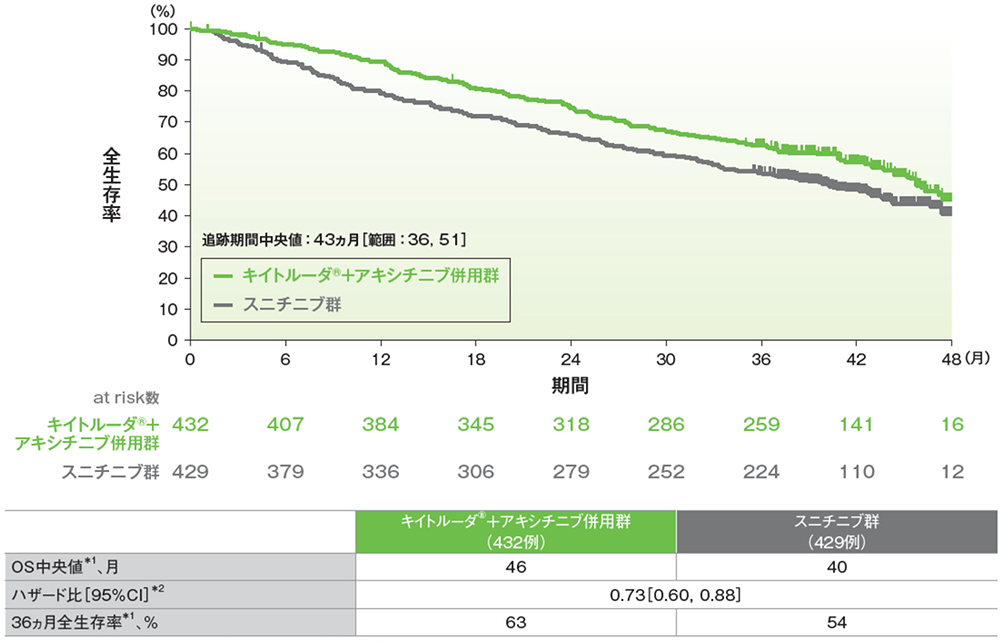
*1 打ち切りデータはproduct-limit (Kaplan-Meier)法に基づく
*2 投与群を共変量とし、IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子(無作為化に用いた層別因子)とした層別Cox比例ハザードモデルに基づく
(追跡期間中央値:43ヵ月、範囲:36, 51)
- 最終解析におけるOS中央値はキイトルーダ®+アキシチニブ併⽤群で46ヵ⽉、スニチニブ群で40ヵ⽉であり、36ヵ⽉時点の全⽣存率はキイトルーダ®+アキシチニブ併⽤群で63%、スニチニブ群で54%でした。
Plimack ER et al. Eur Urol 2023; 84: 449-454 (本試験はMSD社の資⾦提供により⾏われた。)
部分集団因子別の全生存期間:OS(サブグループ解析)
全生存期間(OS)の部分集団因子別ハザード比のフォレストプロット(ITT集団)
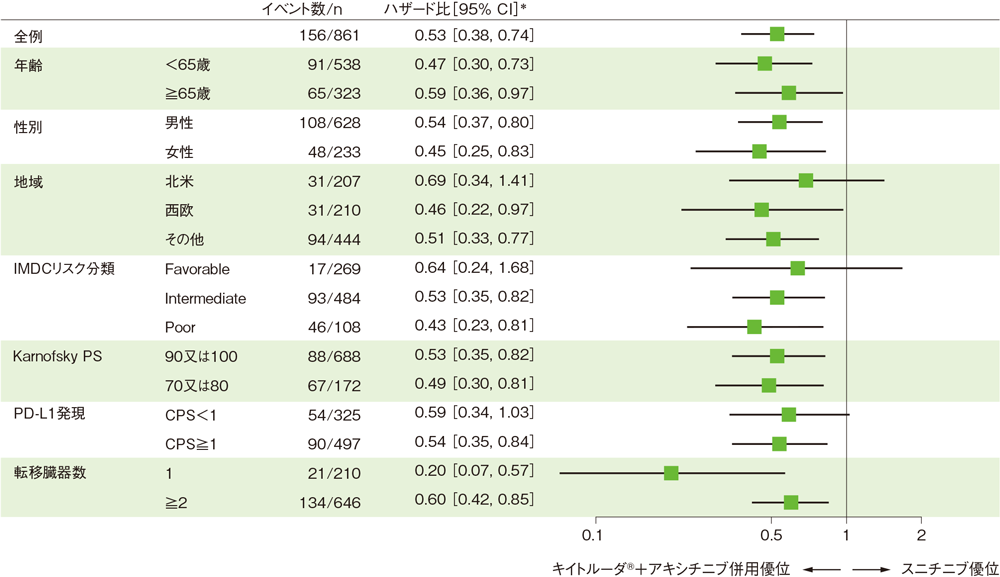
*投与群を共変量とし、IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子とした層別Cox比例ハザードモデルに基づく
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
主要評価項目 無増悪生存期間:PFS
無増悪生存期間(PFS)のKaplan-Meier曲線(ITT集団;検証的解析結果)
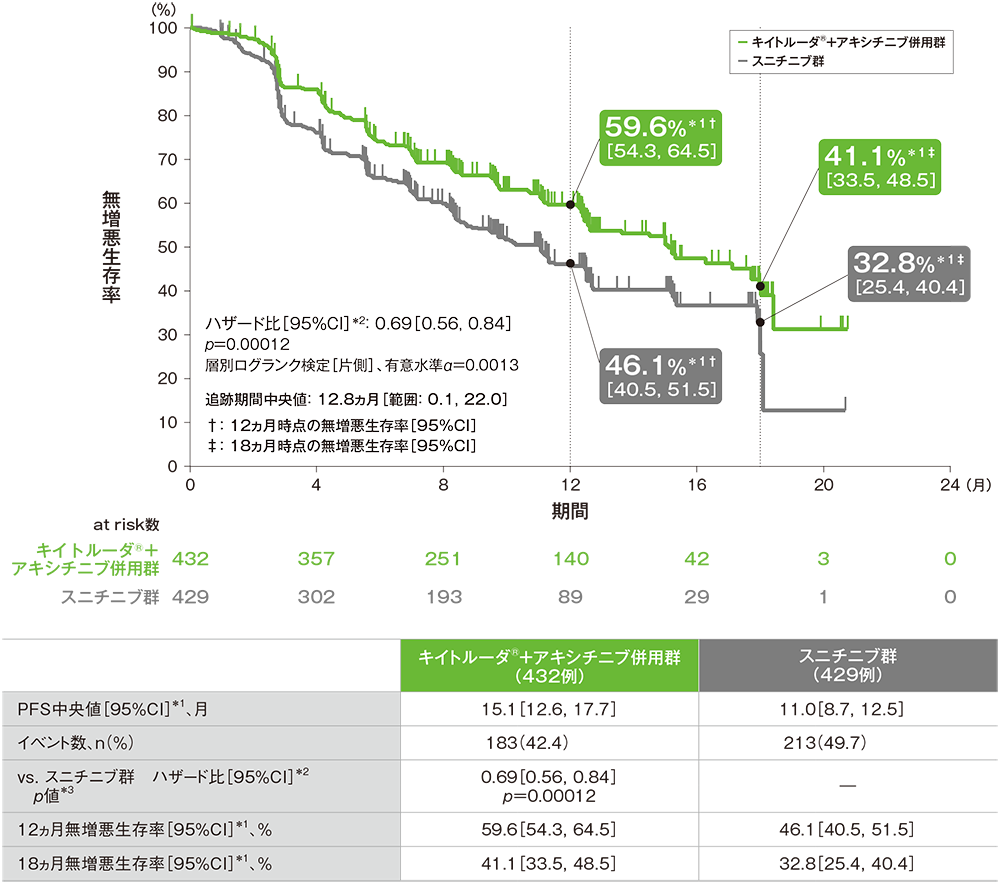
RECISTガイドライン1.1版に基づくBICRによる評価
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子(無作為化に用いた層別因子)とした層別Cox比例ハザードモデルに基づく
*3 層別ログランク検定[片側](層別因子は無作為化に用いた層別因子)、有意水準α=0.0013
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
- 第1回中間解析において、キイトルーダ®+アキシチニブ併用群はスニチニブ群に対して、PFSを有意に延長しました(ハザード比:0.69、95%CI:0.56, 0.84、p=0.00012、層別ログランク検定[片側]、有意水準α=0.0013;検証的解析結果)。
PFS中央値はキイトルーダ®+アキシチニブ併用群で15.1ヵ月(95%CI:12.6, 17.7)、スニチニブ群で11.0ヵ月(95%CI:8.7, 12.5)、12ヵ月時点の無増悪生存率はそれぞれ59.6%(95%CI:54.3, 64.5)、46.1%(95%CI:40.5, 51.5)、18ヵ月時点の無増悪生存率はそれぞれ41.1%(95%CI:33.5, 48.5)、32.8%(95%CI:25.4, 40.4)でした。
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
無増悪⽣存期間(PFS)のKaplan-Meier曲線(ITT集団)
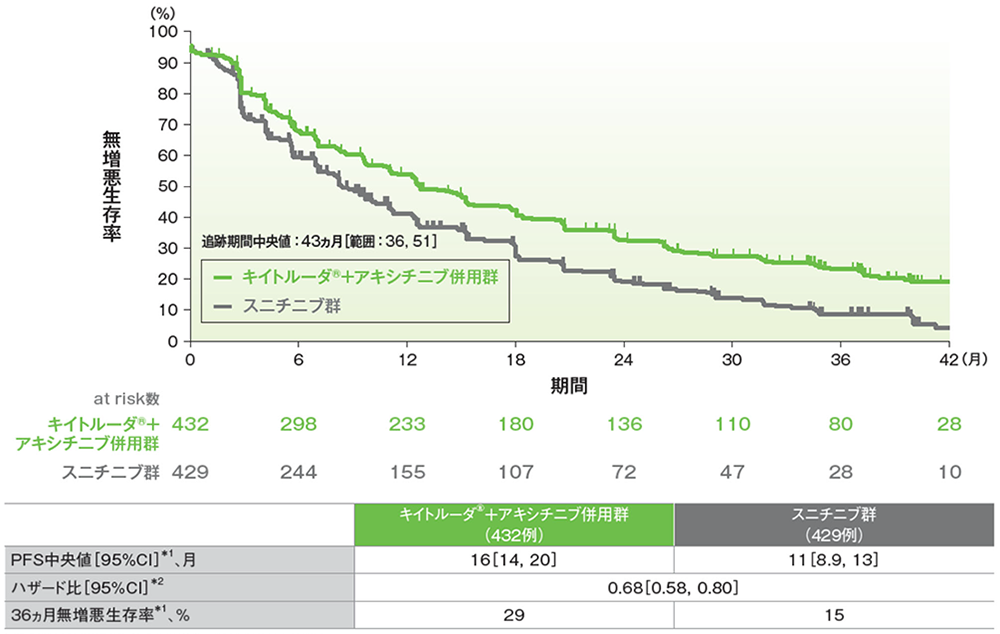
RECISTガイドライン1.1版に基づくBICRによる評価
*1 打ち切りデータはproduct-limit (Kaplan-Meier)法に基づく
*2 投与群を共変量とし、IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子(無作為化に用いた層別因子)とした層別Cox比例ハザードモデルに基づく
(追跡期間中央値:43ヵ月、範囲:36, 51)
- 最終解析におけるPFS中央値はキイトルーダ®+アキシチニブ併⽤群で16ヵ⽉、スニチニブ群で11ヵ⽉であり、36ヵ⽉時点の無増悪⽣存率はキイトルーダ®+アキシチニブ併⽤群で29%、スニチニブ群で15%でした。
Plimack ER et al. Eur Urol 2023; 84: 449-454 (本試験はMSD社の資⾦提供により⾏われた。)
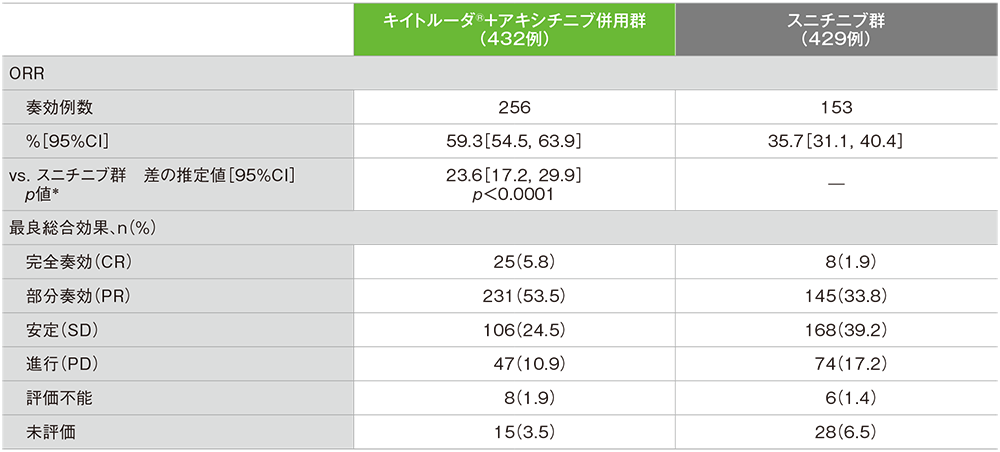
副次評価項目 奏効率:ORR
奏効率(ORR:CR+PR)(ITT集団)
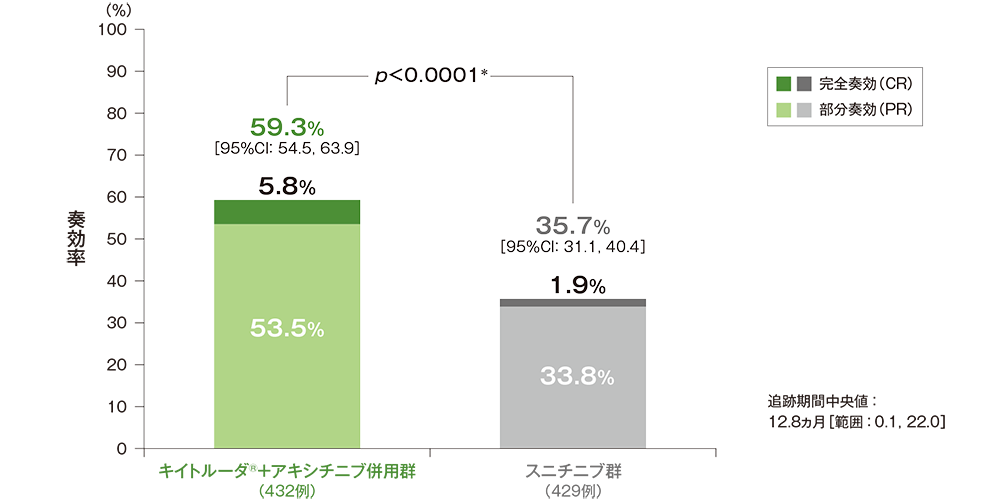
ORR及び最良総合効果(ITT集団)
RECISTガイドライン1.1版に基づくBICRによる評価
*IMDCリスク分類(favorable、intermediate、poor)及び地域(北米、西欧、その他)を層別因子としたMiettinen and Nurminen法[片側]、有意水準α=0.025
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
- 第1回中間解析におけるORRはキイトルーダ®+アキシチニブ併用群59.3%(95%CI:54.5, 63.9)、スニチニブ群35.7%(95%CI:31.1, 40.4)であり、キイトルーダ®+アキシチニブ併用群のORRはスニチニブ群に対して有意に高い値でした(群間差:23.6%、95%CI:17.2, 29.9、p<0.0001、層別Miettinen and Nurminen法 [片側]、有意水準α=0.025)。
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
奏効率(ORR:CR+PR)の要約(ITT集団)
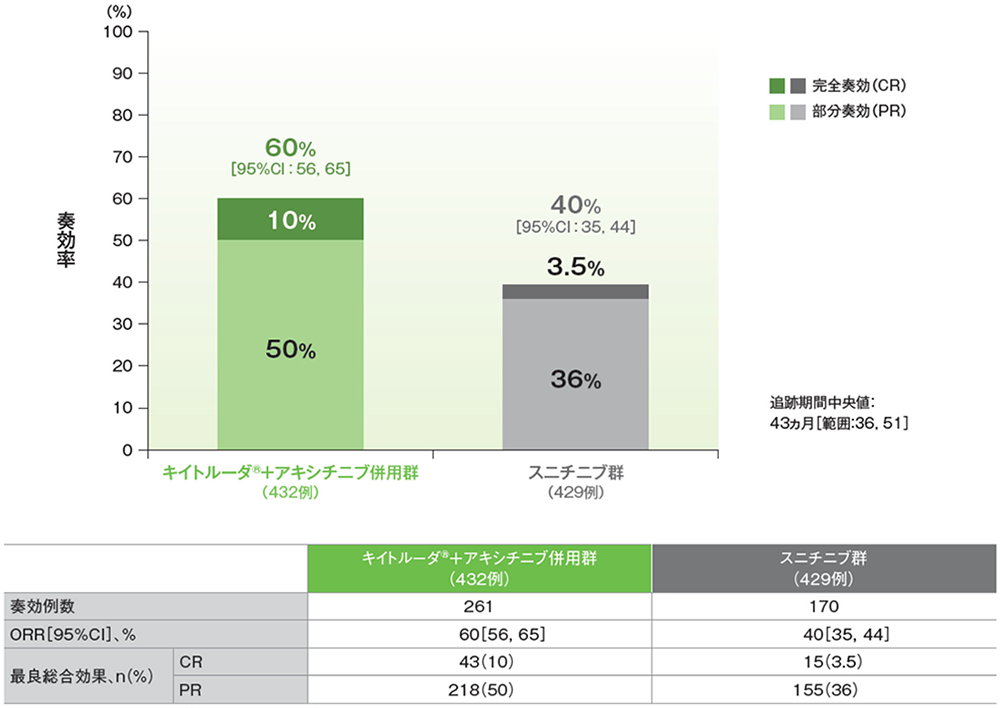
RECISTガイドライン1.1版に基づくBICRによる評価
(追跡期間中央値:43ヵ月、範囲:36, 51)
- 最終解析におけるORRはキイトルーダ®+アキシチニブ併⽤群で60%、スニチニブ群で40%でした。
Plimack ER et al. Eur Urol 2023; 84: 449-454 (本試験はMSD社の資⾦提供により⾏われた。)
副次評価項目 病勢コントロール率:DCR
病勢コントロール率(DCR:CR+PR+6ヵ月以上のSD)(ITT集団)
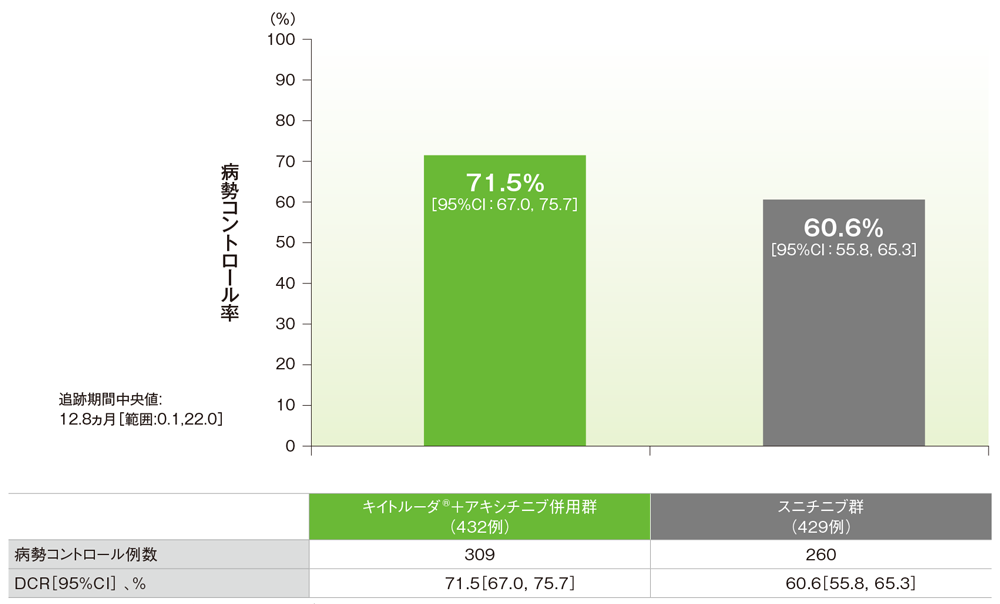
RECISTガイドライン1.1版に基づくBICRによる評価
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
- 第1回中間解析におけるDCRはキイトルーダ®+アキシチニブ併用群で71.5%(95%CI:67.0, 75.7)、スニチニブ群60.6% (95%CI:55.8, 65.3)でした。
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
副次評価項目 奏効期間:DOR
奏効が認められた患者における奏効期間(DOR)のKaplan-Meier曲線(ITT集団)
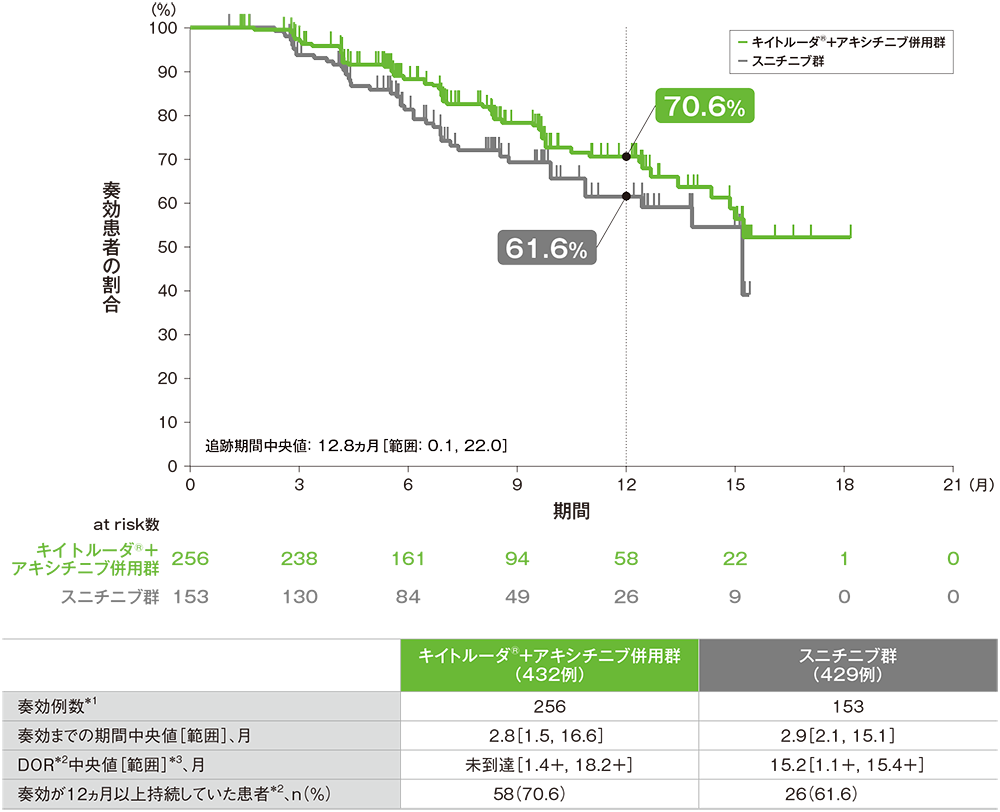
*1 RECISTガイドライン1.1版に基づくBICRによる評価において、完全奏効又は部分奏効を認めた症例数
*2 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*3 「+」は最後の疾患評価からPDがみられないことを示す
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
- 第1回中間解析における奏効までの期間中央値はキイトルーダ®+アキシチニブ併用群で2.8ヵ月(範囲:1.5, 16.6)、スニチニブ群で2.9ヵ月(範囲:2.1, 15.1)でした。
DOR中央値はキイトルーダ®+アキシチニブ併用群で未到達(範囲:1.4+, 18.2+)、スニチニブ群で15.2ヵ月(範囲:1.1+, 15.4+)でした。
奏効が認められた患者のうち、奏効が12ヵ月以上持続した割合は、キイトルーダ®+アキシチニブ併用群で70.6%、スニチニブ群で61.6%でした。
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
安全性
第1回中間解析におけるキイトルーダ®+アキシチニブ併用群の副作用は413/429例(96.3%)に認められました。主な副作用(発現率20%以上)は下痢210例(49.0%)、高血圧179例(41.7%)、甲状腺機能低下症135例(31.5%)、疲労130例(30.3%)、手掌・足底発赤知覚不全症候群119例(27.7%)、アラニンアミノトランスフェラーゼ増加102例(23.8%)、発声障害98例(22.8%)、アスパラギン酸アミノトランスフェラーゼ増加97例(22.6%)、食欲減退94例(21.9%)、悪心91例(21.2%)でした。重篤な副作用は102例(23.8%)に認められ、2例以上にみられた重篤な副作用は下痢8例(1.9%)、アラニンアミノトランスフェラーゼ増加6例(1.4%)、アスパラギン酸アミノトランスフェラーゼ増加及び肺臓炎 各5例(1.2%)、急性腎障害、肝機能異常及び重症筋無力症 各4例(0.9%)、脳血管発作、大腸炎、脱水、肝炎、肝細胞損傷及び肺塞栓症 各3例(0.7%)、副腎機能不全、心房細動、頭痛、肝酵素上昇、高血糖、高血圧、下垂体炎、肝機能検査値上昇、心筋炎、筋炎、手掌・足底発赤知覚不全症候群及びトランスアミナーゼ上昇 各2例(0.5%)でした。副作用によるキイトルーダ®及びアキシチニブの投与中止は27例(6.3%)に認められ、2例以上に発現したキイトルーダ®及びアキシチニブの投与中止に至った副作用はアラニンアミノトランスフェラーゼ増加5例(1.2%)、アスパラギン酸アミノトランスフェラーゼ増加、肝機能異常及び重症筋無力症 各3例(0.7%)、肝炎2例(0.5%)でした。副作用によるキイトルーダ®の投与中止は80例(18.6%)に認められ、2例以上に発現したキイトルーダ®の投与中止に至った副作用はアラニンアミノトランスフェラーゼ増加20例(4.7%)、アスパラギン酸アミノトランスフェラーゼ増加16例(3.7%)、肝機能異常、肝毒性及び重症筋無力症 各4例(0.9%)、肝炎及び急性腎障害 各3例(0.7%)、肝細胞損傷、関節炎、大腸炎、肝機能検査値上昇、心筋炎及び肺臓炎 各2例(0.5%)でした。副作用によるアキシチニブの投与中止は66例(15.4%)に認められ、2例以上に発現したアキシチニブの投与中止に至った副作用はアラニンアミノトランスフェラーゼ増加16例(3.7%)、アスパラギン酸アミノトランスフェラーゼ増加7例(1.6%)、肝機能異常5例(1.2%)、食欲減退及び下痢 各4例(0.9%)、肝炎、重症筋無力症、脳血管発作、手掌・足底発赤知覚不全症候群、蛋白尿及び体重減少 各3例(0.7%)、肺塞栓症及び嘔吐 各2例(0.5%)でした。副作用による死亡は4例(0.9%)で、その内訳は心筋炎、壊死性筋膜炎、重症筋無力症及び肺臓炎 各1例(0.2%)でした。
スニチニブ群の副作用は415/425例(97.6%)に認められました。主な副作用(発現率20%以上)は高血圧184例(43.3%)、下痢175例(41.2%)、手掌・足底発赤知覚不全症候群168例(39.5%)、疲労142例(33.4%)、味覚異常129例(30.4%)、甲状腺機能低下症119例(28.0%)、悪心111例(26.1%)、食欲減退106例(24.9%)、血小板減少症94例(22.1%)、粘膜の炎症90例(21.2%)、口内炎86例(20.2%)でした。重篤な副作用は60例(14.1%)に認められ、2例以上にみられた重篤な副作用は脱水及び無力症 各4例(0.9%)、下痢、低ナトリウム血症、血小板数減少及び血小板減少症 各3例(0.7%)、高血圧、心不全、粘膜の炎症、疲労、悪性新生物進行、膵炎、肺炎及び敗血症 各2例(0.5%)でした。副作用によるスニチニブの投与中止は43例(10.1%)に認められ、2例以上に発現したスニチニブの投与中止に至った副作用は疲労3例(0.7%)、蛋白尿、無力症、精神疲労及び粘膜の炎症 各2例(0.5%)でした。副作用による死亡は7例(1.6%)で、その内訳は急性心筋梗塞、心停止、胃腸出血、劇症肝炎、肺炎、悪性新生物進行及び頭蓋内出血 各1例(0.2%)でした。
主な副作用(いずれかの投与群で発現率5%以上)(ASaT集団)
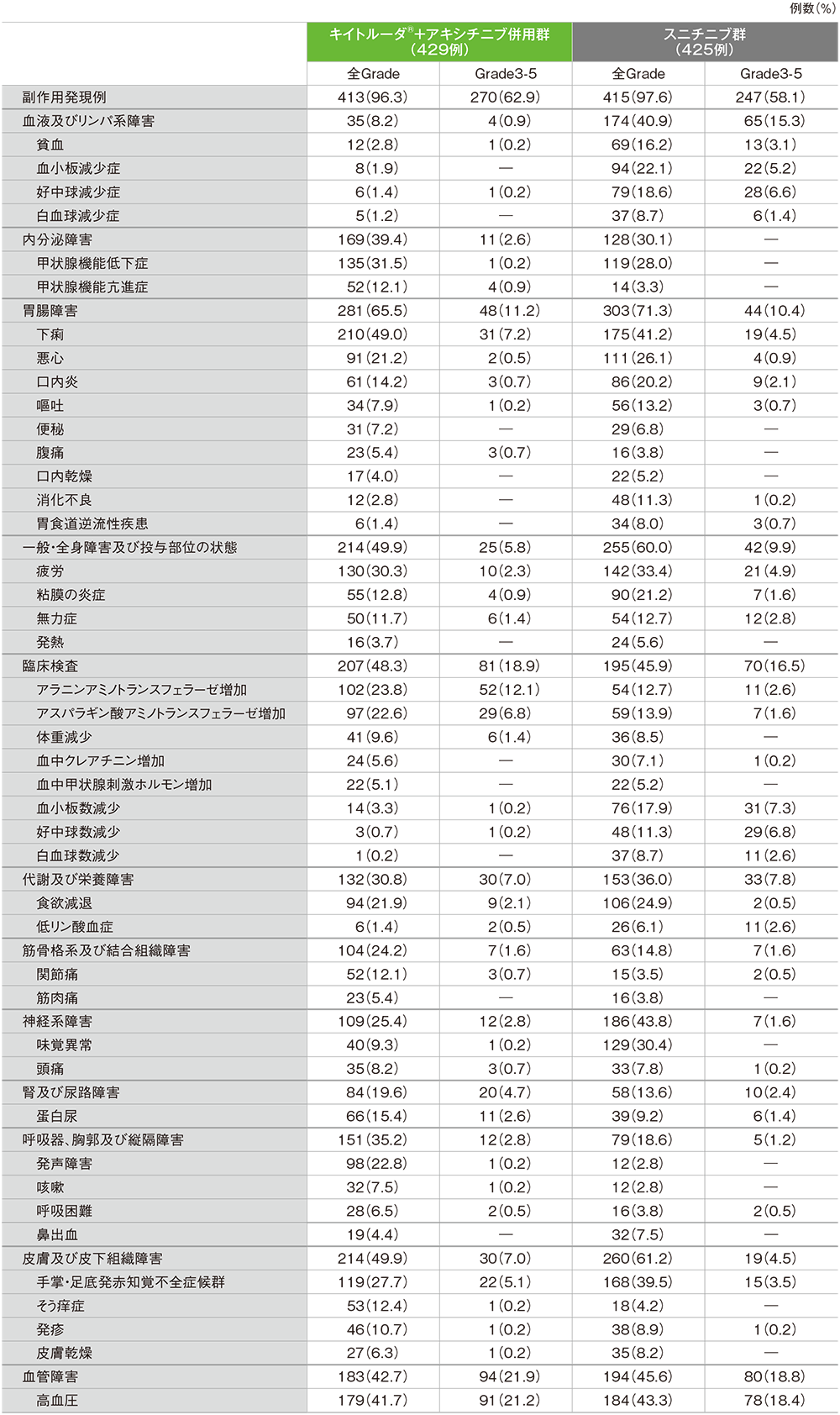
MedDRA/J v21.0、GradeはNCI CTCAE v4.0
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
最終解析における副作⽤は、キイトルーダ®+アキシチニブ併⽤群で429例中413例(96%)、スニチニブ群で425例中415例(98%)に認められました。主な副作⽤(発現率30%以上)は、キイトルーダ®+アキシチニブ併⽤群で下痢231例(54%)、⾼⾎圧191例(45%)、甲状腺機能低下症161例(38%)、疲労140例(33%)、⼿掌・⾜底発⾚知覚不全症候群128 例(30%)、スニチニブ群で⾼⾎圧193例(45%)、下痢188例(44%)、⼿掌・⾜底発⾚知覚不全症候群176例(41%)、疲労151例(36%)、甲状腺機能低下症148例(35%)でした。
副作⽤による死亡はキイトルーダ®+アキシチニブ併⽤群で4例(0.9%;⼼筋炎、重症筋無⼒症、壊死性筋膜炎、肺臓炎 各1例)、スニチニブ群で7例(1.6%;急性⼼筋梗塞、⼼停⽌、胃腸出⾎、肝不全、劇症肝炎、頭蓋内出⾎、肺炎 各1例)に認められました。
重篤な副作⽤及び投与中⽌に⾄った副作⽤は⽂献に記載されていないため、安全性情報の詳細については電⼦添⽂をご参照ください。
主な副作⽤(いずれかの投与群で発現率10%以上)(ASaT集団)
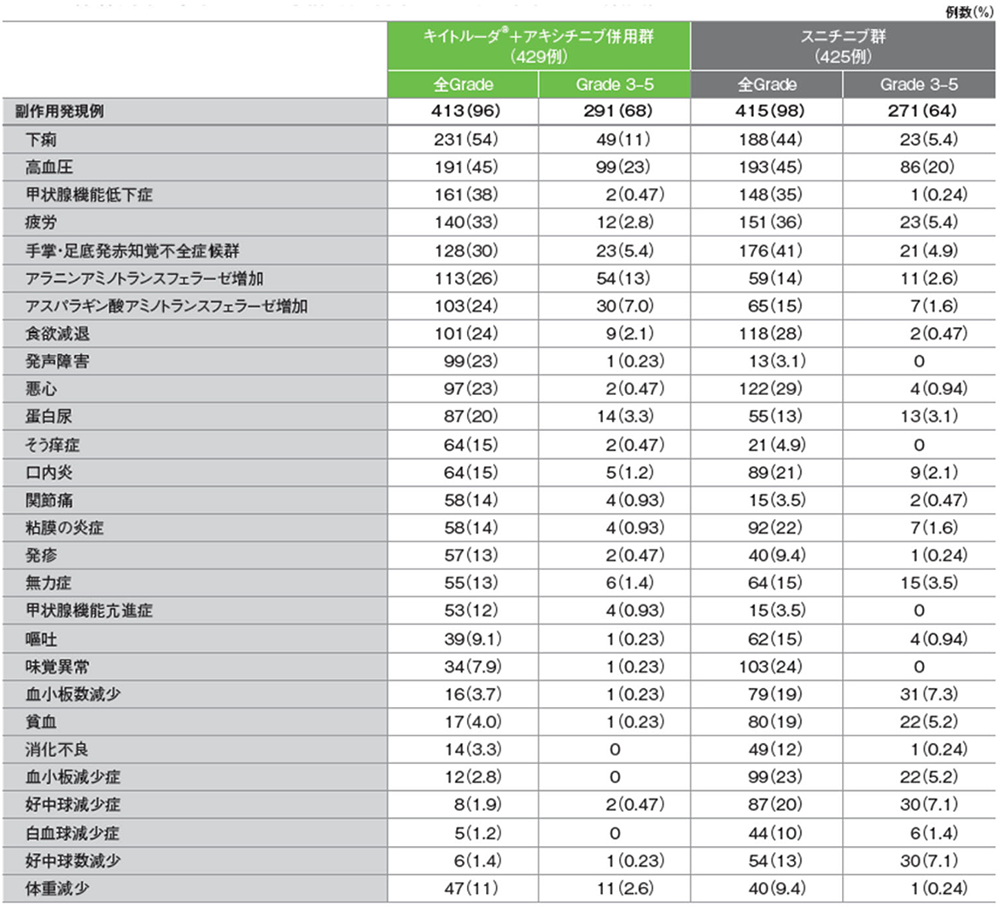
GradeはNCI CTCAE v4.0(追跡期間中央値:43ヵ月、範囲:36, 51)
Plimack ER et al. Eur Urol 2023; 84: 449-454 (本試験はMSD社の資⾦提供により⾏われた。)
免疫関連など特に注目すべき有害事象
第1回中間解析におけるキイトルーダ®+アキシチニブ併用群の免疫関連など特に注目すべき有害事象は220/429例(51.3%)に認められました。主な免疫関連など特に注目すべき有害事象(発現率10%以上)は甲状腺機能低下症152例(35.4%)及び甲状腺機能亢進症55例(12.8%)でした。重篤な免疫関連など特に注目すべき有害事象は42例(9.8%)に認められ、2例以上にみられた重篤な免疫関連など特に注目すべき有害事象は肝炎7例(1.6%)、大腸炎及び肺臓炎 各6例(1.4%)、副腎機能不全5例(1.2%)、筋無力症候群4例(0.9%)、甲状腺機能亢進症、下垂体炎、心筋炎、筋炎、腎炎及び膵炎 各2例(0.5%)でした。免疫関連など特に注目すべき有害事象による投与中止は26例(6.1%)に認められ、2例以上にみられたいずれかの薬剤の投与中止に至った免疫関連など特に注目すべき有害事象は肝炎8例(1.9%)、大腸炎及び筋無力症候群 各4例(0.9%)、肺臓炎3例(0.7%)、心筋炎及び腎炎 各2例(0.5%)でした。免疫関連など特に注目すべき有害事象による死亡は3例(0.7%)に認められ、その内訳は重症筋無力症、肺臓炎及び心筋炎 各1例(0.2%)でした。
スニチニブ群の免疫関連など特に注目すべき有害事象は154/425例(36.2%)に認められました。主な免疫関連など特に注目すべき有害事象(発現率10%以上)は甲状腺機能低下症134例(31.5%)でした。重篤な免疫関連など特に注目すべき有害事象は6例(1.4%)に認められ、2例以上にみられた重篤な免疫関連など特に注目すべき有害事象は甲状腺機能低下症及び膵炎 各2例(0.5%)でした。免疫関連など特に注目すべき有害事象による投与中止は4例(0.9%)に認められ、その内訳は肝炎、Infusion reaction、腎炎及び膵炎 各1例(0.2%)でした。免疫関連など特に注目すべき有害事象による死亡は1例(0.2%)に劇症肝炎を認めました。
免疫関連など特に注目すべき有害事象(ASaT集団)
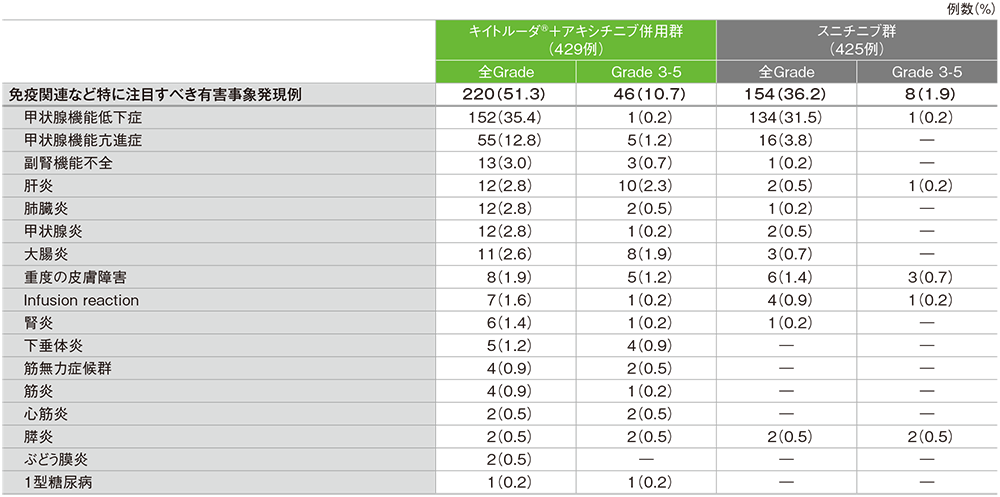
MedDRA/J v21.0、GradeはNCI CTCAE v4.0
(追跡期間中央値:12.8ヵ月、範囲:0.1, 22.0)
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-426試験)
最終解析における免疫関連など特に注⽬すべき有害事象は、キイトルーダ®+アキシチニブ併⽤群で429例中246例(57%)、スニチニブ群で425例中186例(44%)に認められました。主な免疫関連など特に注⽬すべき有害事象(発現率10%以上)は、キイトルーダ®+アキシチニブ併⽤群で甲状腺機能低下症175例(41%)、甲状腺機能亢進症57例(13%)、スニチニブ群で甲状腺機能低下症165例(39%)でした。
免疫関連など特に注⽬すべき有害事象(ASaT集団)
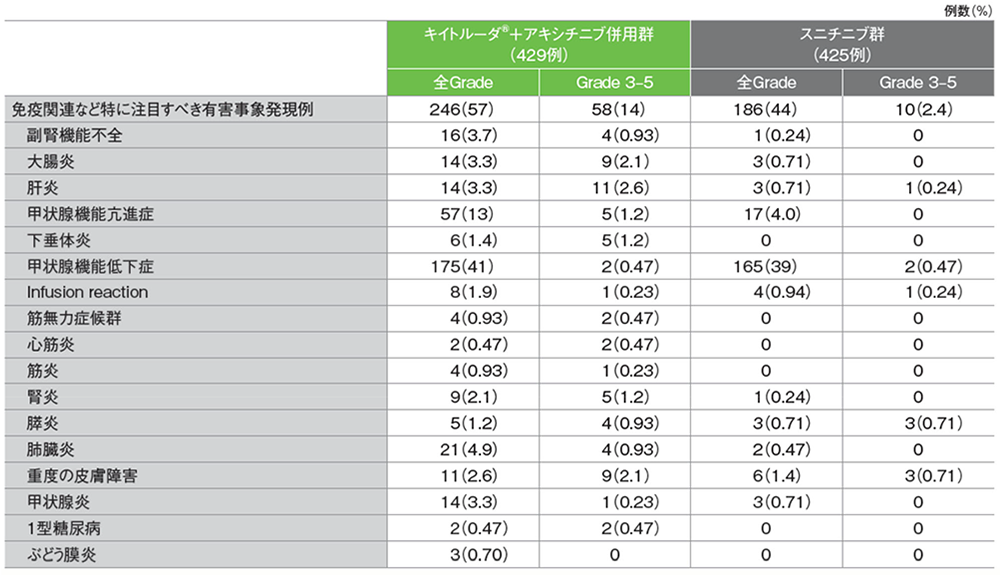
GradeはNCI CTCAE v4.0(追跡期間中央値:43ヵ月、範囲:36, 51)
Plimack ER et al. Eur Urol 2023; 84: 449-454 (本試験はMSD社の資⾦提供により⾏われた。)
キイトルーダ®とアキシチニブ併用療法期間中は、以下の検査スケジュールを参考に、患者さんの状態を観察してください。
キイトルーダ®200mgを3週間間隔及びアキシチニブを開始用量として5mg 1日2回投与したKEYNOTE-426試験における検査スケジュール(キイトルーダ®は最大35回投与)
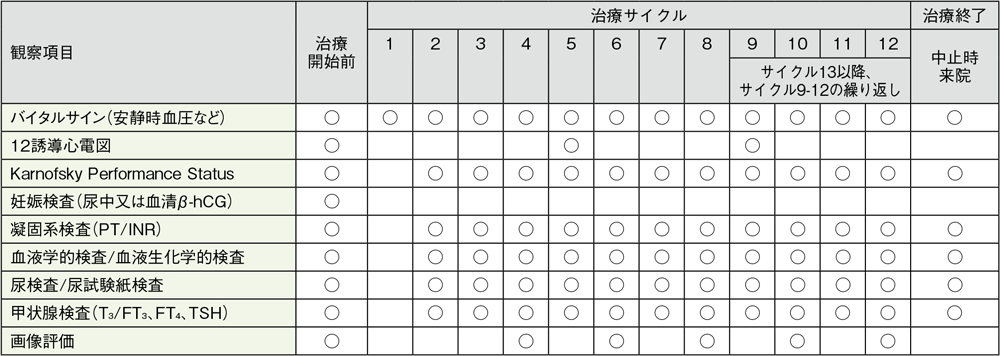
バイタルサイン:体温、脈拍、呼吸数、体重、安静時血圧。1サイクル目は8日目に血圧及び脈拍のみ収集。服用前に毎回血圧を測定し、日誌に記録。
妊娠検査:妊娠の可能性がある女性の場合に、治療開始前72時間以内に実施
血液学的検査:ヘマトクリット、ヘモグロビン、血小板数、白血球数、白血球分画、好中球絶対数、単球絶対数、好酸球絶対数、好塩基球絶対数、リンパ球絶対数
血液生化学的検査:総蛋白、アルブミン、ALT、AST、ALP、総ビリルビン、直接ビリルビン、BUN、クレアチニン、Na、K、Cl、Ca、補正カルシウム(スクリーニング時のみ)、LDH、P、血糖値、HCO3–
尿検査:潜血、糖、蛋白、比重、顕微鏡検査(異常値の場合)
画像評価:12週時及び54週まで6週毎、以降は12週毎