小児<同種造血幹細胞移植> 後期第Ⅱ相国際共同試験(030試験)
臨床成績
本試験は一部承認外の用法及び用量が含まれるデータですが、小児の用法及び用量追加の承認時評価資料であるため掲載します。
小児<同種造血幹細胞移植>
後期第Ⅱ相国際共同試験(030試験)1)
CMV感染及び感染症のリスクのある18歳未満の小児同種造血幹細胞移植患者を対象にプレバイミス®を投与した際の薬物動態、有効性、安全性及び忍容性を評価する単群非盲検後期第Ⅱ相試験
1)用法及び用量追加(小児)承認時評価資料(後期第Ⅱ相国際共同試験:030試験)
試験概要
主要目的:
CMV感染及び感染症のリスクのある、出生時から18歳未満の小児同種造血幹細胞移植(HSCT) 患者を対象に、小児患者におけるプレバイミス®の薬物動態を評価する。
対象:
出生時から18歳未満の初回同種HSCT患者※1 65例(うち日本人は5例)
年齢グループ
- 12歳~18歳未満:28例(うち日本人は2例)
- 2歳~12歳未満:29例(うち日本人は3例)
- 出生時~2歳未満:8例
※1:CMV感染及び感染症のリスクがあり、組み入れ前5日以内に採取した検体でCMV DNAが検出されなかった患者を主な選択基準とした。
試験デザイン:
国際多施設共同単群非盲検試験
投与方法:
本試験では3つの年齢グループ(年齢グループ1:12歳~18歳未満、年齢グループ2:2歳~12歳未満、年齢グループ3:出生時~2歳未満)に患者を組み入れた。
患者は、移植後28日までにプレバイミス®の投与を開始し※2、1日1回経口(錠又は顆粒※3)又は静脈内(点滴静注)投与にて移植後14 週(約100日)まで継続した。プレバイミス®の用量は、年齢、体重区分及びシクロスポリン併用の有無に応じて、下表の通りとした。

※2:投与は経口剤で開始することが望ましく、12歳~18歳未満には錠剤を投与する(錠剤の嚥下を嫌がる又は嚥下できない患者には顆粒剤の使用を許容する)ことが望ましいとした。注射剤は、嚥下不能又は経口剤の吸収を妨げる可能性のある状態(例:嘔吐、下痢、吸収不良状態)にのみ使用可能とし、静脈内投与の期間は原則4週間以内とした。
患者が嚥下可能になった場合又は静脈内投与を必要とする状態が解消した場合は、速やかに(次回の投与時に)静脈内投与から経口投与に切り替えることとした。
※3:顆粒剤は、軟らかい食品に混ぜて、又は経鼻/胃瘻チューブを介して投与した。
■プレバイミス®の用量
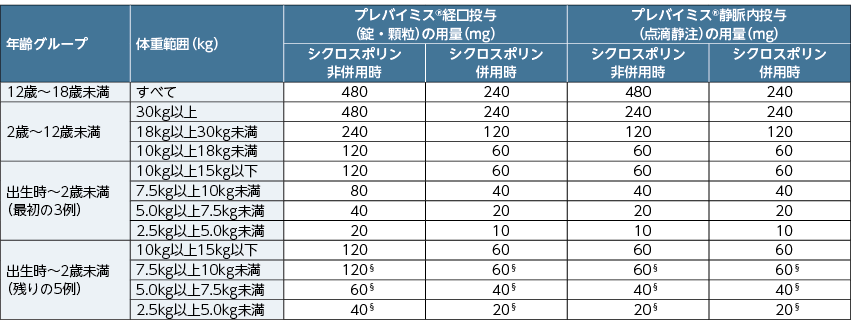
初回投与時の用量は、割付け時に記録された体重を用いて決定された。用量は、治験スケジュールに従って測定された直近の体重に基づき調整した。
§:出生時~2歳未満の年齢グループの最初の3例の薬物動態の評価を受けて、以後の組み入れは体重が10kg未満の患者での用量を増量した。
評価項目:
【主要評価項目】 レテルモビルの薬物動態評価(AUC0-24hr 等)
【副次評価項目】 有効性:移植後14週及び24週以内に臨床的に意味のあるCMV感染※4がみられた患者の割合
安全性:有害事象 等
※4:臨床的に意味のあるCMV感染は以下のように定義した。
「臓器障害を伴うCMV感染症の発症」もしくは「CMV血症の確認及び患者の臨床状態に基づく抗CMV薬による先制治療の開始」
解析計画:
- PKの解析対象集団は、測定可能なPK評価用の検体が1つ以上あり、治験実施計画書に記載されたPK評価の要件を満たすすべての患者[PP(Per protocol)]とした。AUC0-24hr等を算出し、幾何平均及び95%信頼区間(CI)を年齢グループ及び用量別に提⽰した。必要に応じて、AUC0-24hr等の個々の値を年齢グループ及び用量別にプロットした。ノンコンパートメントPK解析にはPK評価用の頻回採血が行われ、頻回採血日にPK評価用検体をすべて採取した患者から構成されたPP集団の部分集団(36例)を用いた。
- 有効性の主要解析対象集団は、治験薬を1回以上投与され、1日目にCMV DNAが検出されなかったすべての患者[FAS(Full analysis set)]とした。患者の割合の95%信頼区間(CI)は、Clopper and Pearson法を用いて算出した。
- 主要な欠測データは非完了例=無効例(NC=F:Non-Completer=Failure)アプローチとし、臨床的に意味のあるCMV感染がみられた患者と本試験を早期中止した又は評価時点(14週又は24週)での有効性に関する測定値が欠測の患者は無効例とみなした。また、年齢区分(12歳~18歳未満、2歳~12歳未満、出生時~2歳未満)ごとの予防投与効果の一貫性を評価するため、サブグループ解析として年齢グループ別に移植後14週及び24週以内の有効性を評価した。
- 安全性の解析対象集団は治験薬を1回以上投与されたすべての患者[APaT(All participants as treated)]とした。
- 安全性の主要解析では、治験薬最終投与後28日までの安全性データを要約した。重篤な副作用及び死亡に至った重篤な有害事象は移植後48週まで収集した。
解析対象例数:
薬物動態解析対象例数(PP):61例(最終的な解析対象例数は60例)(12歳~18歳未満27例、2歳~12歳未満25例、出生時~2歳未満8例)
有効性解析対象例数(FAS):56例(12歳~18歳未満25例、2歳~12歳未満24例、出生時~2歳未満7例)
安全性解析対象例数(APaT):63例(12歳~18歳未満28例、2歳~12歳未満27例、出生時~2歳未満8例)
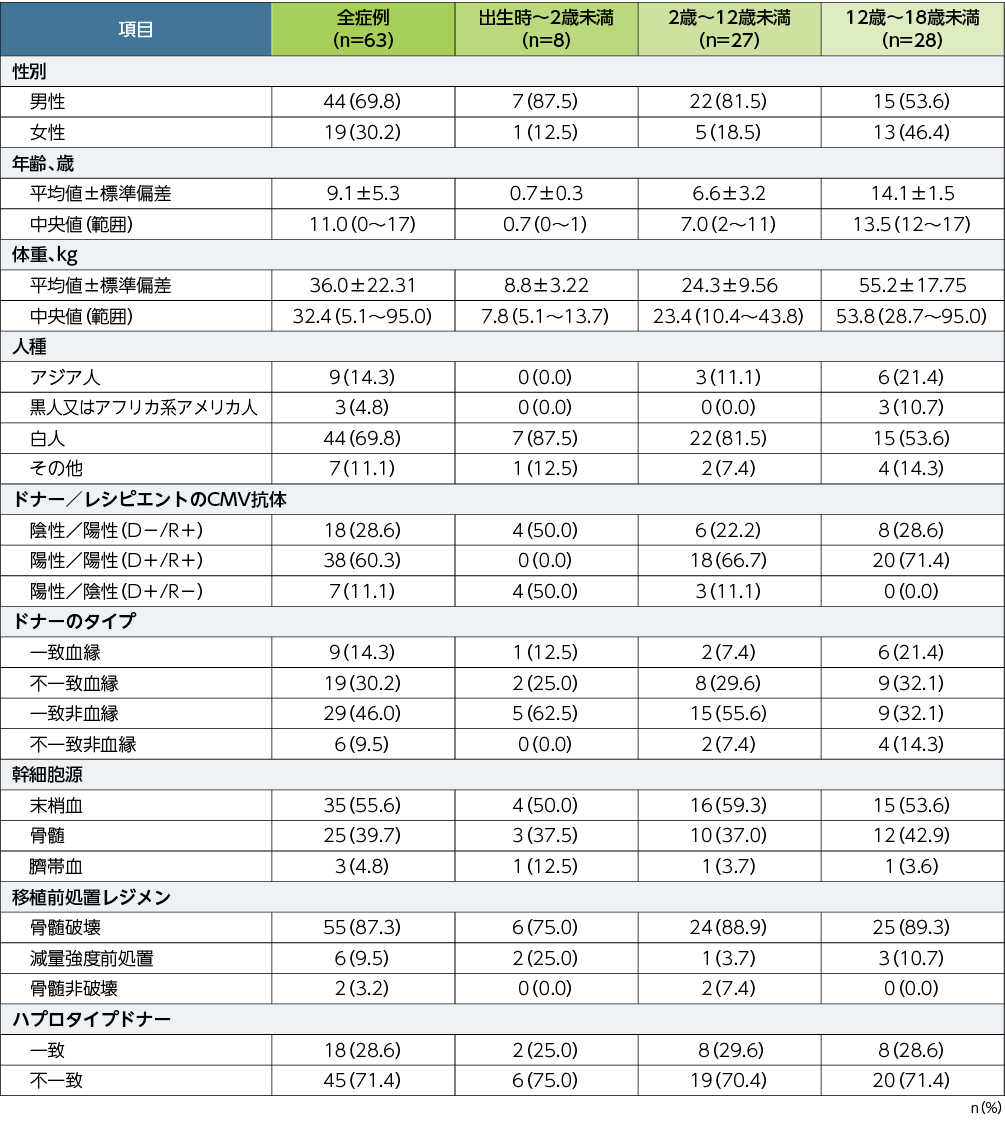
患者背景
■患者背景(APaT)
1. 年齢グループごとのプレバイミス®のAUC0-24hr(主要評価項目)
出生時~18歳未満の患者におけるプレバイミス®の曝露量は、以下の通りでした。
■小児同種造血幹細胞移植患者にプレバイミス®を1日1回経口(錠又は顆粒)又は静脈内投与した際の年齢グループごとの血漿中レテルモビルのAUC0-24hr(ノンコンパートメント解析)(主要評価項目、PP部分集団)
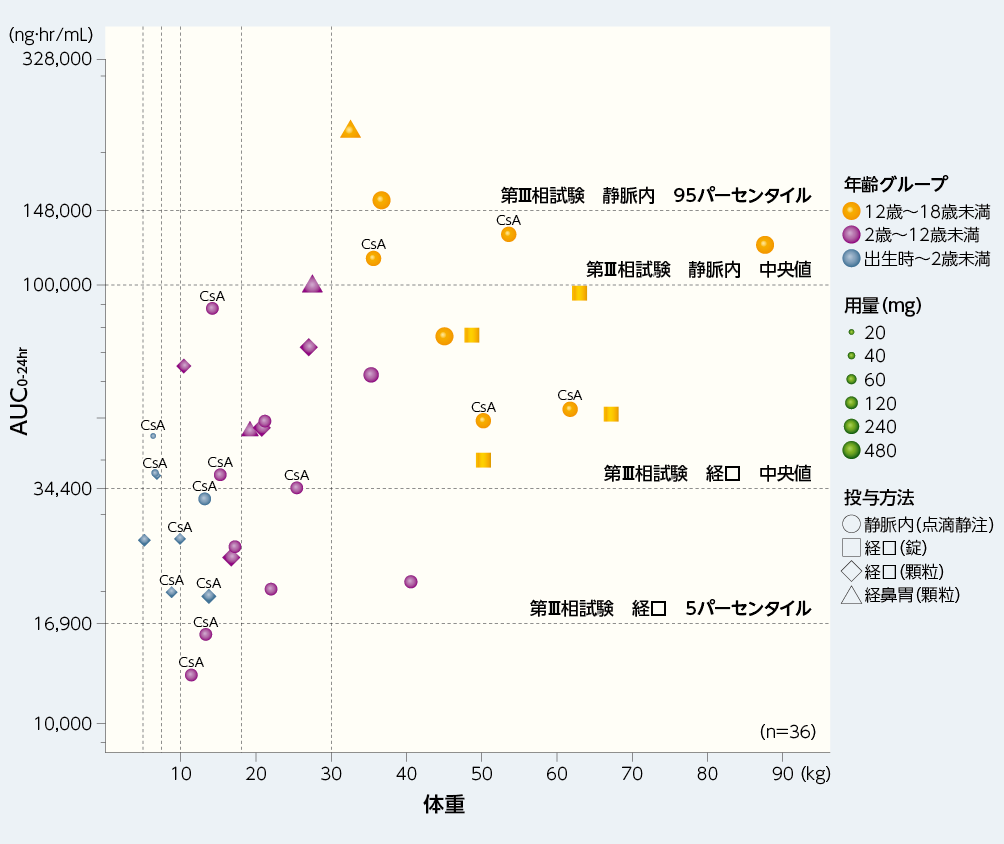
AUC0-24hr:投与間隔あたり(投与後0~24時間まで)の濃度-時間曲線下面積、CsA:シクロスポリン
シンボルの上の「CsA」は、シクロスポリンと併用してプレバイミス®が投与されたことを⽰す。
縦の点線は出生時~2歳未満及び2歳~12歳未満の体重区分を⽰す(12歳~18歳未満の体重の境界はない)。
成人同種造血幹細胞移植患者にプレバイミス®480mgを経口又は静脈内単独投与(シクロスポリン非併用)した際の定常状態での曝露量(AUC0-24hr)の中央値を小児患者における目標曝露量(34,400~100,000ng·hr/mL)とし、プレバイミス®経口投与時の曝露量の5パーセンタイル及び静脈内投与時の曝露量の95パーセンタイルをそれぞれ小児患者における目標曝露量の範囲の下限及び上限(16,900~148,000ng·hr/mL)とした。プレバイミス®の第Ⅰ相試験で観察された最も高い曝露量を許容可能な曝露量の上限(328,000ng·hr/mL)とした。
参考:薬物動態(母集団薬物動態解析:日本人及び外国人データ)2)
2)社内資料(後期第Ⅱ相臨床試験データを用いた母集団薬物動態(PPK)解析、小児同種造血幹細胞移植患者)
030試験データに基づき、小児同種造血幹細胞移植患者の母集団薬物動態解析が実施され、小児における用法及び用量が⽀持されました。
■小児同種造血幹細胞移植患者にプレバイミス®を1日1回経口又は静脈内投与した際の定常状態における予測AUC0-24hr
経口投与(錠又は顆粒)
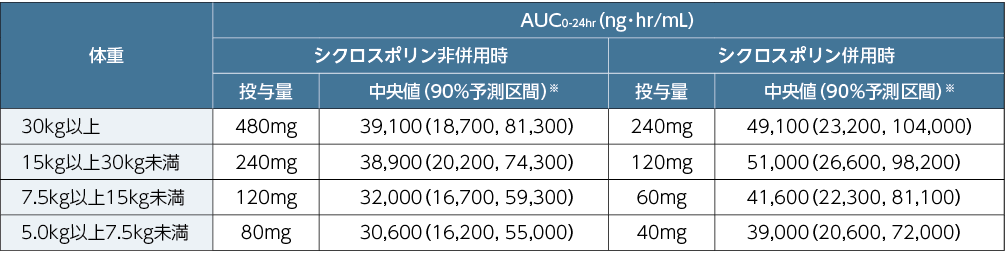
静脈内投与
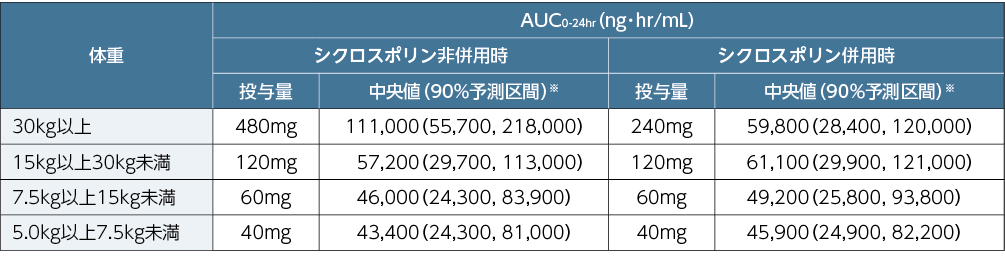
※:中央値及び90%予測区間は、小児同種造血幹細胞移植患者の母集団薬物動態モデルを用いた個体間変動を考慮したシミュレーションに基づき算出した。
2. 移植後14週及び24週以内に臨床的に意味のあるCMV感染※1がみられた患者の割合(副次評価項目、サブグループ解析)
移植後14週及び24週以内に臨床的に意味のあるCMV感染がみられた患者(NC=Fアプローチ※2)の割合は、それぞれ19.6%(11/56例)及び25.0%(14/56例)でした。各年齢グループ別の割合は以下の通りでした。
■移植後14週及び24週以内に臨床的に意味のあるCMV感染がみられた患者の割合(全症例:副次評価項目、各年齢グループ:サブグループ解析、FAS)
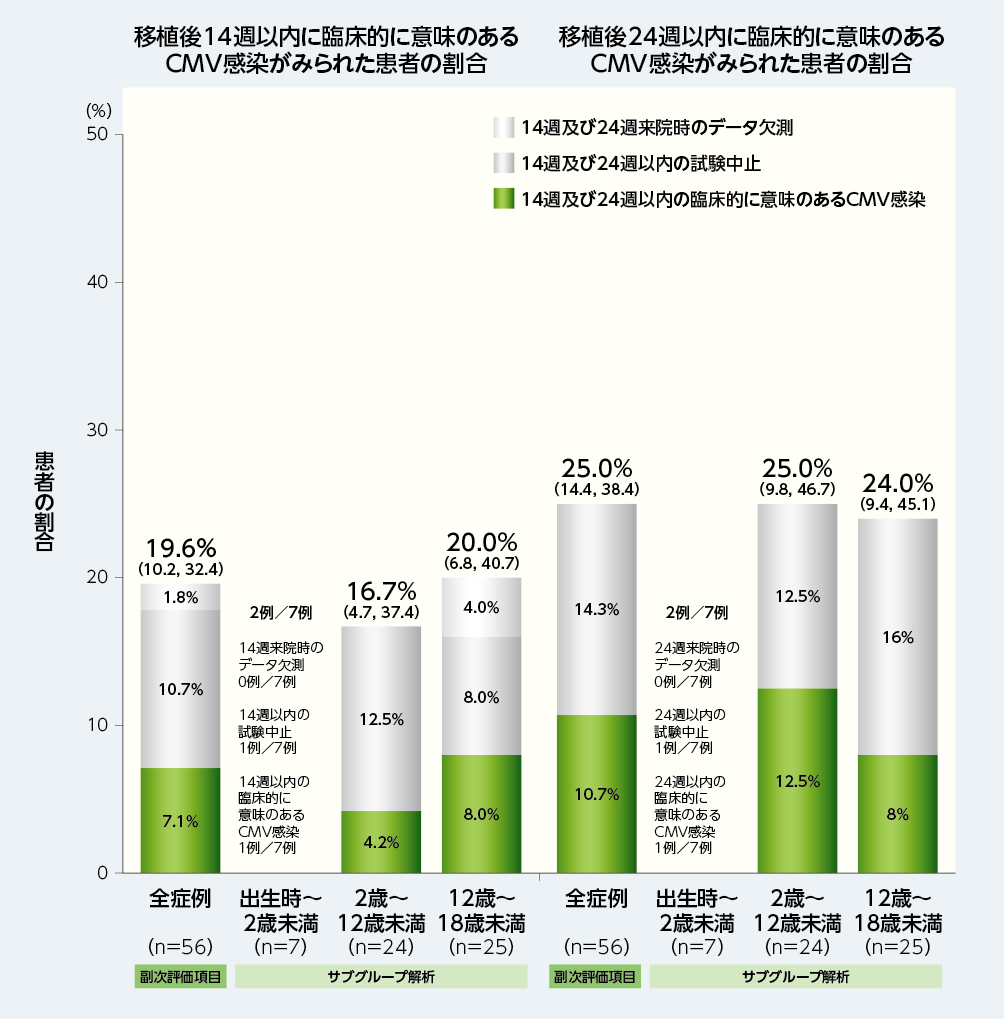
図内の数値は、患者の割合(95%CI)を⽰した。95%CIはClopper and Pearson法を用いて算出された。
※1:臨床的に意味のあるCMV感染は、「臓器障害を伴うCMV感染症の発症」もしくは「CMV血症の確認及び患者の臨床状態に基づく抗CMV薬による先制治療の開始」と定義した。
※2:NC=F(Non-Completer=Failure)アプローチ:臨床的に意味のあるCMV感染がみられた患者と本試験を早期中止した又は評価時点(14週又は24週)での有効性に関する測定値が欠測の患者は無効例とみなした。
3. 安全性(副次評価項目)
副作用※発現率
移植後14週までの副作用は、63例中20例(31.7%)に認められました。頻度の高かった副作用(発現率3%以上)は、嘔吐11例(17.5%)、悪心及び免疫抑制剤濃度増加各2例(3.2%)でした。重篤な副作用は、12歳~18歳未満で心房細動及び血中ビリルビン増加が各1例でした。投与中止に至った副作用は、12歳~18歳未満で心房細動及び併用薬の薬物濃度治療量以下が各1例でした。本試験において、死亡に至った副作用は認められませんでした。
■後期第Ⅱ相国際共同試験(030試験)における副作用発現率(副次評価項目、APaT)
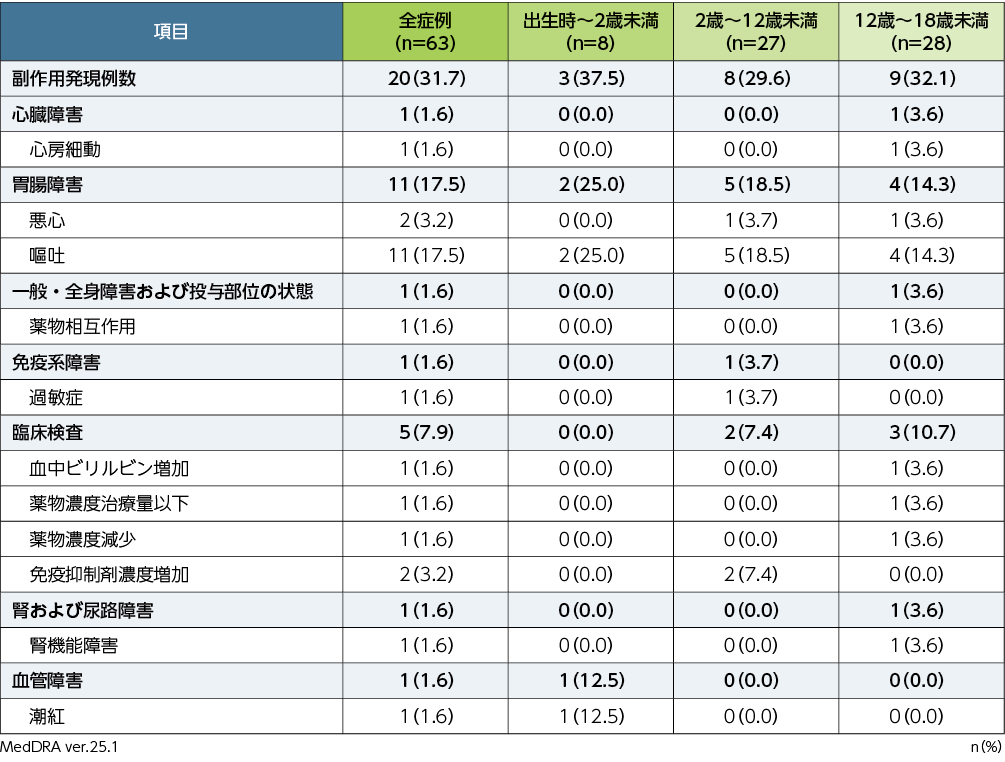
※:治験担当医師が盲検下で治験薬との因果関係ありと判定した有害事象を副作用と定義した。
6. 用法及び用量
・錠240mg 顆粒分包20mg・120mg
通常、成人にはレテルモビルとして480mg(240mg錠2錠又は120mg顆粒4包)を1日1回経口投与する。シクロスポリンと併用投与する場合にはレテルモビルとして240mg(240mg錠1錠又は120mg顆粒2包)を1日1回経口投与する。
通常、小児にはレテルモビルとして以下の用量を1日1回経口投与する。
・点滴静注240mg
通常、成人にはレテルモビルとして480mgを1日1回、約60分かけて点滴静注する。シクロスポリンと併用投与する場合にはレテルモビルとして240mgを1日1回、約60分かけて点滴静注する。
通常、小児にはレテルモビルとして以下の用量を1日1回、約60分かけて点滴静注する。