血糖値低下効果(イプラグリフロジン追加投与試験)
イプラグリフロジン追加投与試験(国内第Ⅲ相検証的試験)
血糖値低下効果(副次評価項目)[24週時]
イプラグリフロジン追加投与群およびプラセボ追加投与群の治療期24週時のベースラインからの空腹時血糖値変化量(最小二乗平均[95%信頼区間])は、それぞれ−30.3mg/dL[−35.5, −25.0]および−2.1mg/dL[−7.6, 3.3]でした。両群の群間差(最小二乗平均[95%信頼区間])は、−28.1mg/dL[−34.8, −21.5]であり、空腹時血糖値変化量はプラセボ追加投与群に対してイプラグリフロジン追加投与群で有意に大きいことが示されました(p<0.001、cLDAモデル)。
イプラグリフロジン追加投与群およびプラセボ追加投与群の治療期24週時のベースラインからの食後2時間血糖値変化量(最小二乗平均[95%信頼区間])は、それぞれ−52.4mg/dL[−61.5, −43.2]および−3.8mg/dL[−13.3, 5.7]でした。両群の群間差(最小二乗平均[95%信頼区間])は、−48.5mg/dL[−59.6, −37.5]であり、食後2時間血糖値変化量はプラセボ追加投与群に対してイプラグリフロジン追加投与群で有意に大きいことが示されました(p<0.001、cLDAモデル)。
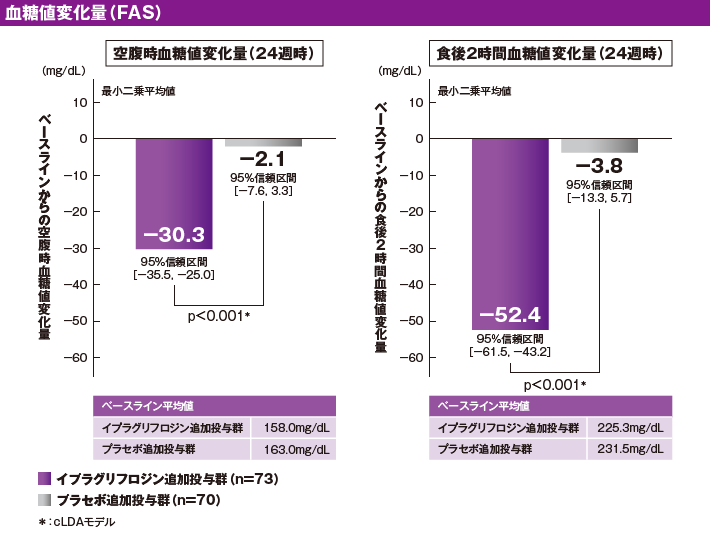
国内第Ⅲ相臨床試験P843(承認申請時評価資料)
安全性:副作用発現率は、イプラグリフロジン追加投与群が11.0%(8/73例)、プラセボ追加投与群が5.7%(4/70例)であった。イプラグリフロジン追加投与群で発現した副作用は、便秘および頻尿が2.7%(2/73例)、血圧低下、体重減少、脱水、筋痙縮、頚動脈狭窄、脳梗塞、頭痛および陰部そう痒症がいずれも1.4%(1/73例)、プラセボ追加投与群で発現した副作用は、便秘2.9%(2/70例)、筋痙縮および湿疹がいずれも1.4%(1/70例)であった。なお、死亡例は認められなかった。重篤な副作用は、イプラグリフロジン追加投与群では1.4%(1/73例:脳梗塞1例)に認められ、プラセボ追加投与群では認められなかった。投与中止に至った副作用は、イプラグリフロジン追加投与群で2.7%(2/73例:脳梗塞1例、頻尿および頭痛1例)に認められたが、プラセボ追加投与群には認められなかった。
試験概要
目 的 :食事・運動療法に加えシタグリプチン50mg単剤治療で血糖コントロールが不十分な日本人2型糖尿病患者に、イプラグリフロジン50mgまたはプラセボを1日1回24週間追加投与したときのプラセボを対照とした併用投与の有効性を検証するとともに、安全性および忍容性を検討する。
対 象 :20歳以上で、食事・運動療法に加えシタグリプチン50mg1日1回単剤治療で血糖コントロールが不十分な日本人2型糖尿病患者(HbA1cが7.0%以上10.0%以下)143例(イプラグリフロジン追加投与群73例、プラセボ追加投与群70例)
観察期間は、2型糖尿病に対する前治療に基づき被験者ごとにAグループ(10週間)またはBグループ(2週間)のいずれかが割り当てられた。
方 法 :プラセボ観察期を除くスクリーニング期および観察期に非盲検下にてシタグリプチン50mgを1日1回朝、最大10週間経口投与した後、プラセボ観察期として、シタグリプチンと同時に、イプラグリフロジン50mgに対応するプラセボを1日1回朝、2週間経口投与した。プラセボ観察期終了後、イプラグリフロジン追加投与群(イプラグリフロジン50mgを追加投与)またはプラセボ追加投与群(イプラグリフロジン50mgに対応するプラセボを追加投与)に無作為に割り付け、二重盲検下で1日1回朝、24週間経口投与した(治療期)。朝投与はすべて食前食後を問わないものとした。
評価項目:
<主要評価項目>
治療期24週時のベースラインからのHbA1c変化量(検証項目)
<副次評価項目>
(1) 治療期24週時のベースラインからの空腹時血糖値変化量
(2) 治療期24週時のベースラインからの食後2時間血糖値変化量
(3) 治療期24週時のベースラインからのグルコース合計AUC0-2hr(食事負荷時)変化量
(4) 治療期24週時のベースラインからの体重変化量
<三次評価項目>
治療期24週時のHbA1c(7.0%未満)達成割合
<安全性>
有害事象(低血糖を含む)、バイタルサイン(体重、血圧、脈拍)、臨床検査、心電図
解析計画:主要評価項目である治療期24週時のベースラインからのHbA1c変化量の群間差を、cLDAモデル*を用いて推定および検定し、事前に規定された群間差(イプラグリフロジン追加投与群-プラセボ追加投与群)が約-0.3%以下である場合に、主要仮説であるプラセボに対する優越性が検証されたとした。また、副次評価項目についても同様にcLDAモデル*を用いて解析した。
*cLDAモデル(constrained Longitudinal Data Analysis):制約つき経時データ解析
本解析では、ベースラインの平均値は投与群間で共通で、ベースライン後の各時点では投与群ごとに平均値が異なると仮定し、投与群、時点、血糖降下薬による治療歴の有無、投与群と時点の交互作用、時点と血糖降下薬による治療歴の有無の交互作用、投与群と時点と血糖降下薬による治療歴の有無の交互作用、ベースラインの推算糸球体濾過量(eGFR)をモデルに入れて、共分散分析を行った。
主要な有効性解析対象集団はFAS、安全性解析対象集団はASaTとした。
FAS(Full Analysis Set):有効性評価に関する最大の解析対象集団
ASaT(All Subjects as treated):治療期用治験薬を1回以上投与されたすべての被験者
「禁忌」等その他項目はこちらをご参照ください。