小児侵襲性肺炎球菌感染症予防における0歳児での初回免疫の重要性
本動画では、小児侵襲性肺炎球菌感染症予防の観点から、0歳児における初回免疫での免疫獲得の重要性について、詳しくご説明いたします。
承認時評価資料:国内第Ⅲ相試験(033試験)
Suzuki H, et al. Vaccine. 2023;41(34):4933-4940. [利益相反:本試験はMSD社の支援により行われた。
著者の一部はMSD社の社員である。]
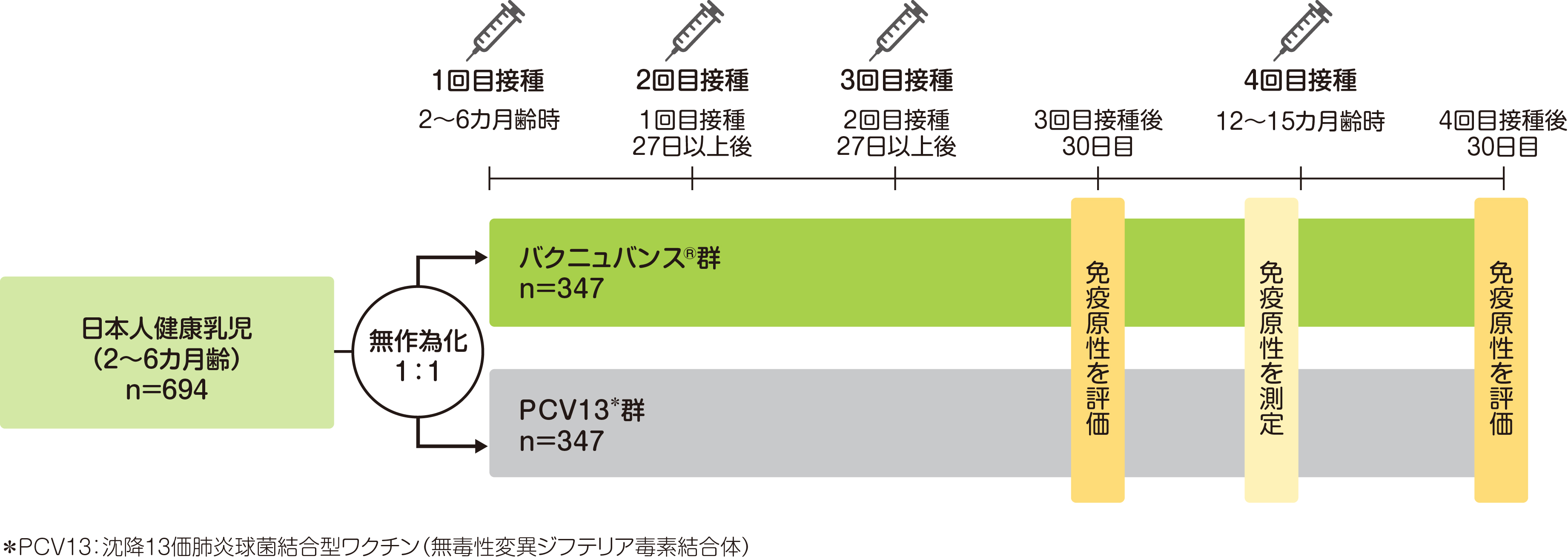
| 試験 デザイン | 無作為化、実薬対照、多施設共同、二重盲検試験 |
| 目的 | 日本人健康乳児(2~6カ月齢)を対象に、 バクニュバンス®の安全性、忍容性、および免疫原性について検討する。 |
| 対象 | 日本人健康乳児(2~6カ月齢)694例(バクニュバンス®群347例、PCV13群347例) |
| 方法 | バクニュバンス®群またはPCV13群に月齢(2カ月齢、3カ月齢および4~6カ月齢)で層別し、1:1の比で無作為に割り付け、初回免疫として27日間以上の間隔をあけてバクニュバンス®またはPCV13 0.5mLを3回皮下接種後、追加免疫として12~15カ月齢時に1回皮下接種し、3、4回目の接種後30日目に免疫原性を評価した。なお、他の小児用ワクチンとの同時接種は医師の判断で可能とした。 |
| 評価項目 免疫原性 | 主要評価項目:3回目接種後30日目のPCV13と共通の13血清型に対する血清型特異的IgG抗体保有率、バクニュバンス®固有の2血清型(22Fおよび33F)に対する血清型特異的IgG抗体保有率、 PCV13と共通の13血清型に対する血清型特異的IgG GMC* 副次評価項目:3回目接種後30日目のバクニュバンス®固有の2血清型に対する血清型特異的IgG GMC、4回目接種後30日目のPCV13と共通の13血清型に対する血清型特異的IgG抗体保有率、バクニュバンス®固有の2血清型に対する血清型特異的IgG抗体保有率など |
| 評価項目 安全性 | 主要評価項目:バクニュバンス®接種後1~14日目までの事前に規定した注射部位の有害事象(発赤/紅斑、腫脹、圧痛/疼痛および硬結)、バクニュバンス®接種後1~14日目までの事前に規定した全身性の有害事象(易刺激性、傾眠/傾眠状態、食欲減退および蕁麻疹/膨疹)、試験終了までの重篤な副反応 その他の評価項目:バクニュバンス®およびPCV13接種後1~7日目に体温を測定した。 |
* GMC:幾何平均抗体濃度
| 解析計画 免疫原性 | 免疫原性の解析には治験実施計画書を逸脱していない全ての被験者(PP集団*)を用いた。 主要評価項目:以下、検証項目として3項目設定した。 ① PCV13と共通の13血清型の3回目接種後30日目の血清型特異的IgG抗体保有率(血清型特異的IgG抗体濃度≧0.35μg/mLの被験者割合)を求め、PCV13群に対するバクニュバンス®群の非劣性※1を検証した。 ②3回目接種後30日目のバクニュバンス®固有の2血清型について、PCV13群の血清型特異的IgG抗体保有率が最も低い共通血清型に対するバクニュバンス®群の非劣性※1を検証した。 ③ PCV13と共通の13血清型の3回接種後30日目の血清型特異的IgG GMC比を求め、PCV13群に対するバクニュバンス®群の非劣性※2を検証した。 血清型特異的IgG抗体保有率の群間差(バクニュバンス®群-PCV13群)、および月齢で層別したMiettinen & Nurminen法で95%信頼区間(CI)を算出した。Cochran-Mantel-Haenszel法による重みを用いて、層で調整した割合の群間差を算出した。 血清型特異的IgG GMCを自然対数変換し、接種群および月齢を要因とする分散分析モデルを用いて解析した。自然対数尺度での群間差(バクニュバンス®群-PCV13群)および95%CIをこのモデルより算出し、元の尺度に戻してIgG GMC比(バクニュバンス®群/PCV13群)を求めた。 ※1 非劣性基準:血清型特異的IgG抗体保有率の差(バクニュバンス®群-PCV13群)の両側95%CIの下限>-10%(片側p値<0.025) ※2 非劣性基準:血清型特異的IgG GMC比(バクニュバンス®群/PCV13群)の両側95%CIの下限>0.5(片側p値<0.025) |
| 解析計画 安全性 | 安全性解析対象集団は、ワクチンを接種した全ての被験者(APaT集団**)を用いた。安全性および忍容性は、有害事象およびバクニュバンス®またはPCV13接種後の体温の測定値を含む全ての安全性評価項目を用いて、臨床的な観点から検討した。 |
* PP(Per-protocol)集団:治験実施計画書を逸脱していない全ての被験者
** APaT(All Participants as Treated)集団:ワクチンを接種した全ての被験者
無作為化、実薬対照、多施設共同、二重盲検試験
主要評価項目の「3回接種後30日目のPCV13 と共通する血清型における血清型特異的IgG抗体保有率」では、バクニュバンス®群は、PCV13共通の13血清型において、いずれの血清型においてもPCV13群に対する非劣性が検証されました。
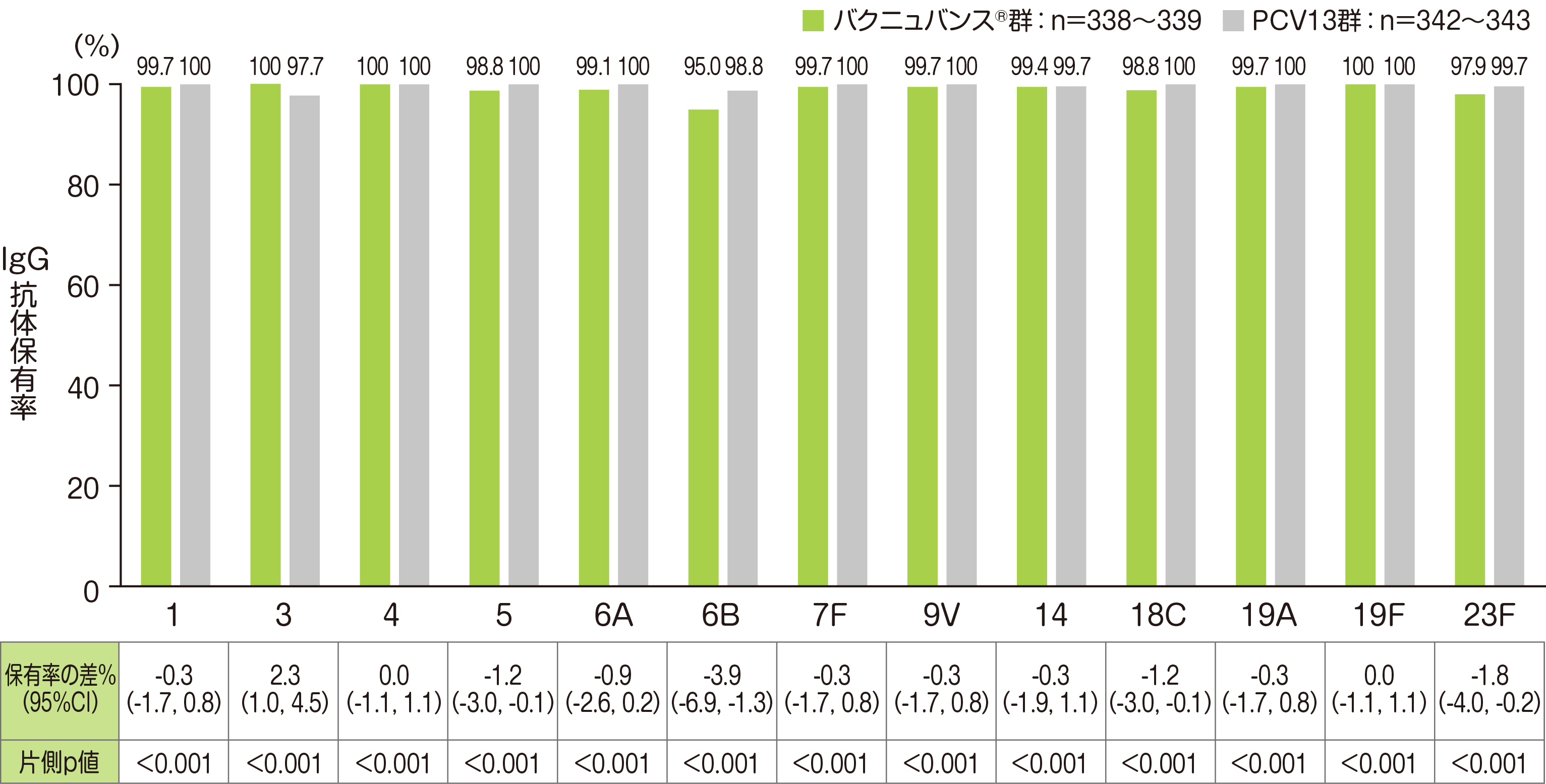
• それぞれの血清型において解析可能な例数は異なる。治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用い、本評価において、解析可能な例数を用いて集計を行った。
• 非劣性基準:血清型特異的IgG抗体保有率の差(バクニュバンス®群-PCV13群)の両側95%CIの下限>-10%(片側p値<0.025)
• Cochran-Mantel-Haenszel法による層別割合の差を算出し、Miettinen & Nurminen法(層別因子:月齢)で95%CI、p値を算出した。
※ 血清型特異的IgG抗体保有率:血清型特異的IgG抗体濃度≧0.35μg/mLの被験者割合
2つ目の主要評価項目である、「3回目接種後30日目のバクニュバンス®固有の2血清型における血清型特異的IgG抗体保有率」については、バクニュバンス®固有の血清型である22F、33FのIgG抗体保有率を、PCV13群で最も低い値を示した「血清型3」と比較しました。
その結果、血清型22Fでは、PCV13群の血清型特異的IgG抗体保有率が最も低い値を示した血清型3に対する非劣性が検証されました。血清型33Fは非劣性基準を満たさず、非劣性は検証されませんでした。
しかし、バクニュバンス®群の33Fに対する血清型特異的IgG抗体保有率は90.9%でした。
![【主要評価項目】 3回目接種後30日目のバクニュバンス®固有の2血清型に対する血清型特異的IgG抗体保有率※[PCV13の血清型特異的IgG抗体保有率が最も低い共通血清型(血清型3) との比較]](https://www.msdconnect.jp/wp-content/uploads/sites/5/2024/03/14.jpg)
• 免疫原性の解析には、治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用いた。
• 非劣性は、バクニュバンス®固有の2血清型とPCV13の血清型特異的IgG抗体保有率が最も低い共通血清型(血清型3)との比較に基づいて検証した。
• 非劣性基準:血清型特異的IgG抗体保有率の差(バクニュバンス®群-PCV13群)の両側95%CIの下限>-10%(片側p値<0.025)
• Cochran-Mantel-Haenszel法による層別割合の差を算出し、Miettinen & Nurminen法(層別因子:月齢)で95%CI、p値を算出した。
※ 血清型特異的IgG抗体保有率:血清型特異的IgG抗体濃度≧0.35μg/mLの被験者割合
3つ目の主要評価項目である、「3回目接種後30日目のPCV13と共通する血清型の血清型特異的IgG GMC」では、PCV13と共通の13血清型において、3回目接種後30日目の血清型特異的IgG GMCはPCV13群に対する非劣性が検証されました。
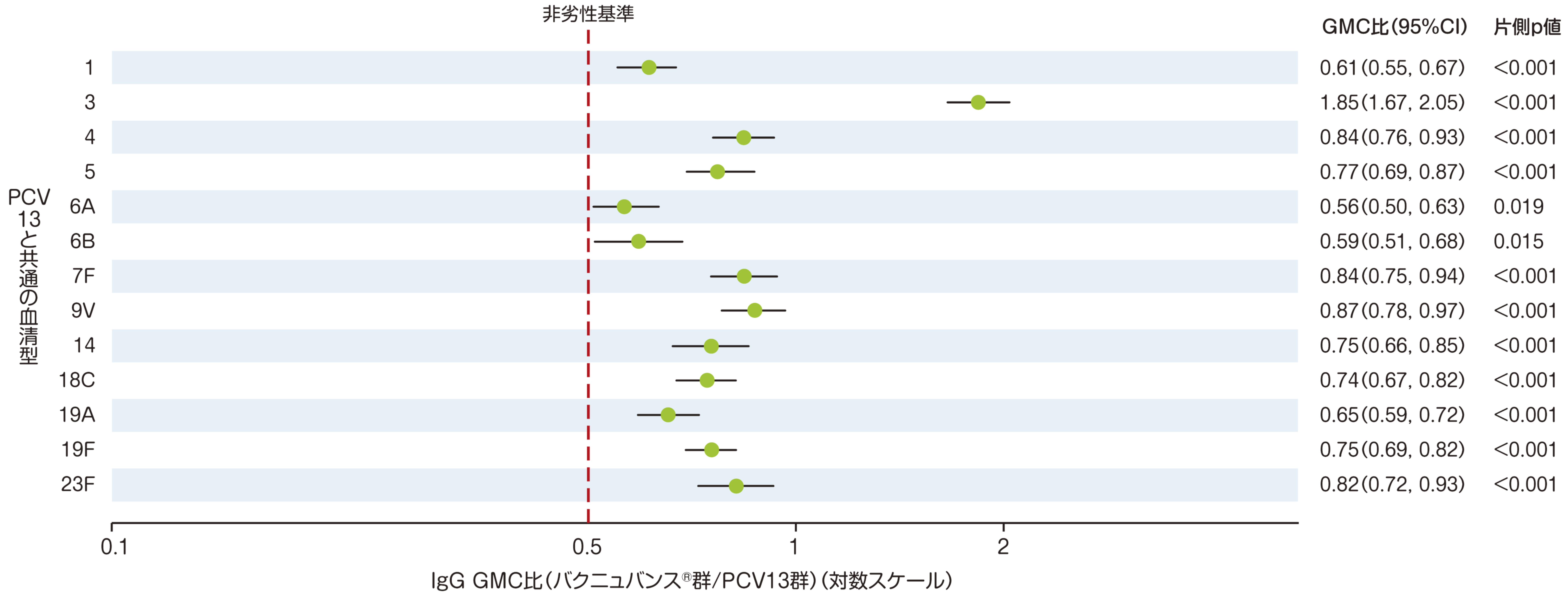
• バクニュバンス®群:n=338~339、PCV13群:n=342~343
• それぞれの血清型において解析可能な例数は異なる。治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用い、本評価において、解析可能な例数を用いて集計を行った。
• 血清型特異的IgG GMC比(95%CI)
• 非劣性基準:血清型特異的IgG GMC比(バクニュバンス®群/PCV13群)の両側95%CIの下限>0.5(片側p値<0.025、分散分析モデル)
* GMC:幾何平均抗体濃度
副次評価項目である「バクニュバンス®固有の2血清型に対する血清型特異的IgG GMC」は、3回目接種後30日目の血清型特異的IgG GMC比において、血清型22Fで107.45、血清型33Fで32.48でした。
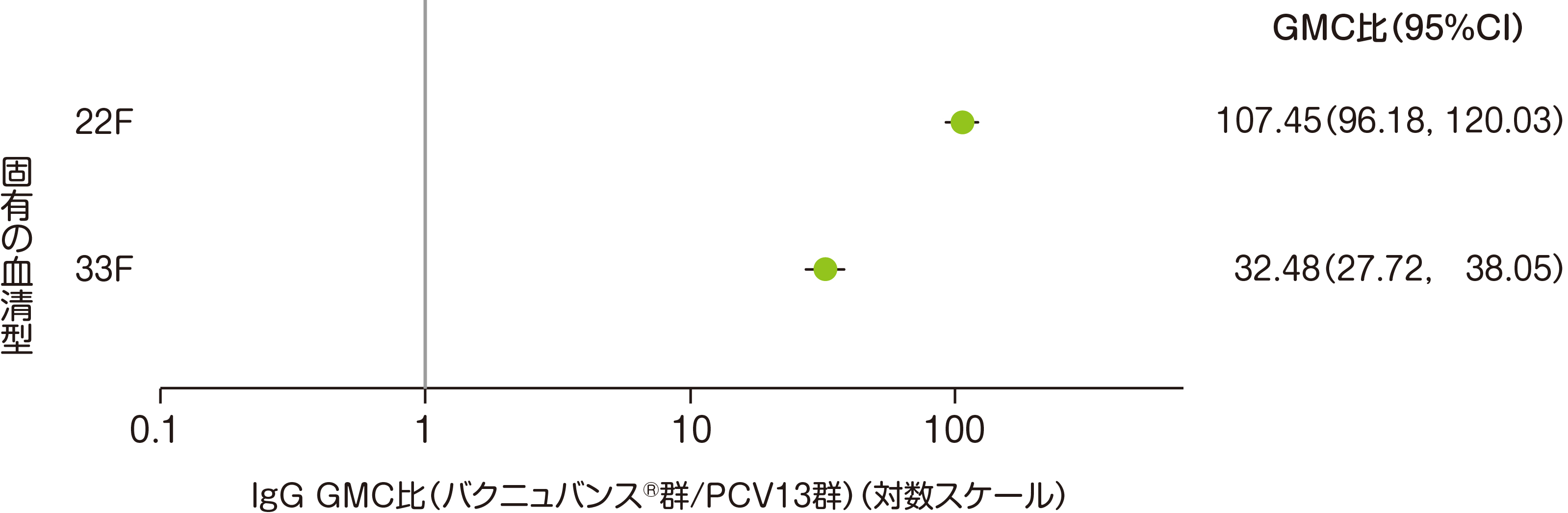
• バクニュバンス®群:n=339、PCV13群:(22F)n=343、(33F)n=337
• 治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用いた。
• 血清型特異的IgG GMC比(95%CI)
• 分散分析モデル
* GMC:幾何平均抗体濃度
副次評価項目である「4回目接種後30日目のPCV13と共通の13血清型に対する血清型特異的IgG抗体保有率」は、バクニュバンス®はPCV13共通の13血清型において、4回目接種後30日目の血清型特異的IgG抗体保有率は、血清型1、4、23Fで99.7%、それ以外の血清型は100%でした。
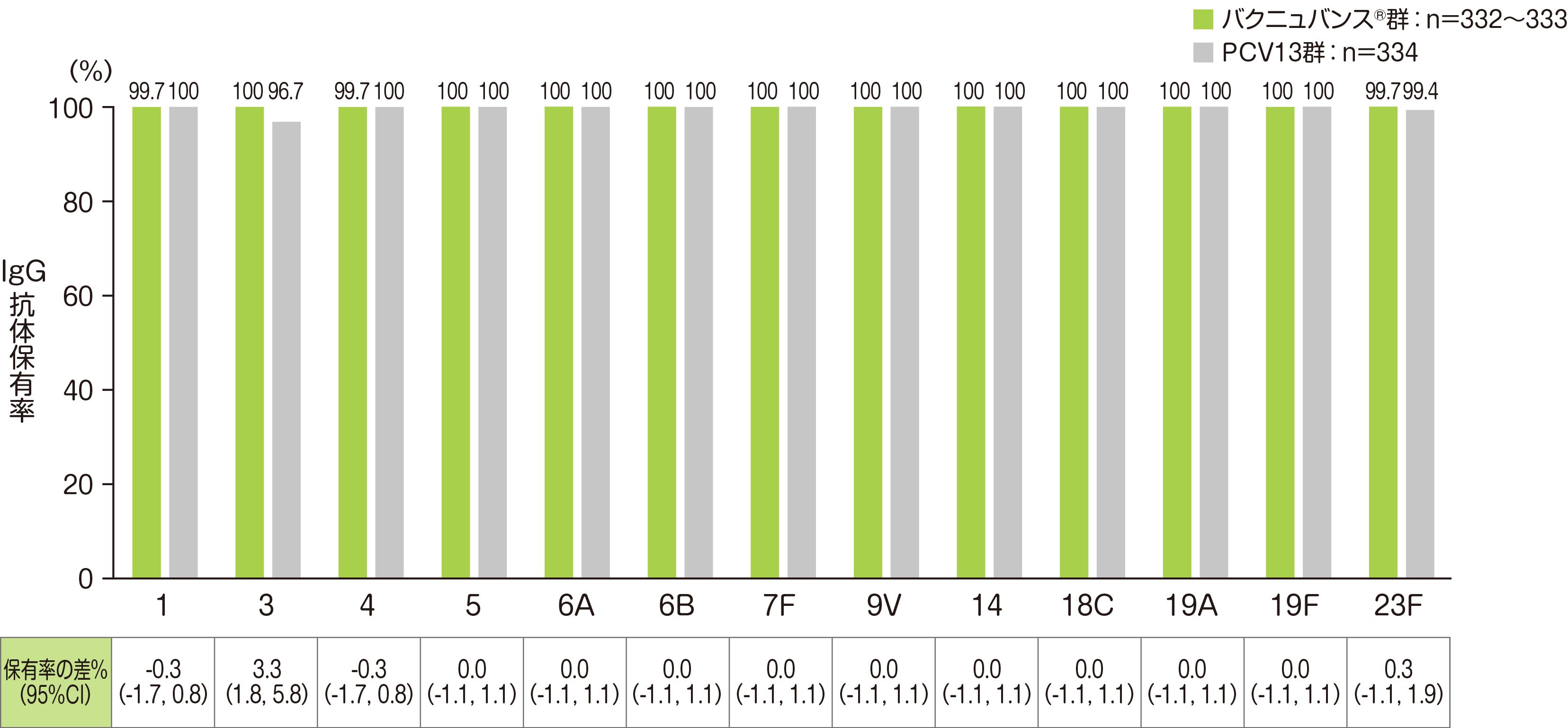
• バクニュバンス®群の血清型において解析可能な例数は異なる。治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用い、本評価において、解析可能な例数を用いて集計を行った。
• Cochran-Mantel-Haenszel法による層別割合の差を算出し、Miettinen & Nurminen法(層別因子:月齢)で95%CIを算出した。
※ 血清型特異的IgG抗体保有率:血清型特異的IgG抗体濃度≧0.35μg/mLの被験者割合
副次評価項目である「4回目接種後30日目のバクニュバンス®固有の2血清型に対する血清型特異的IgG抗体保有率」については、4回目接種後30日目の血清型22F、33Fの血清型特異的IgG抗体保有率はいずれも100%でした。
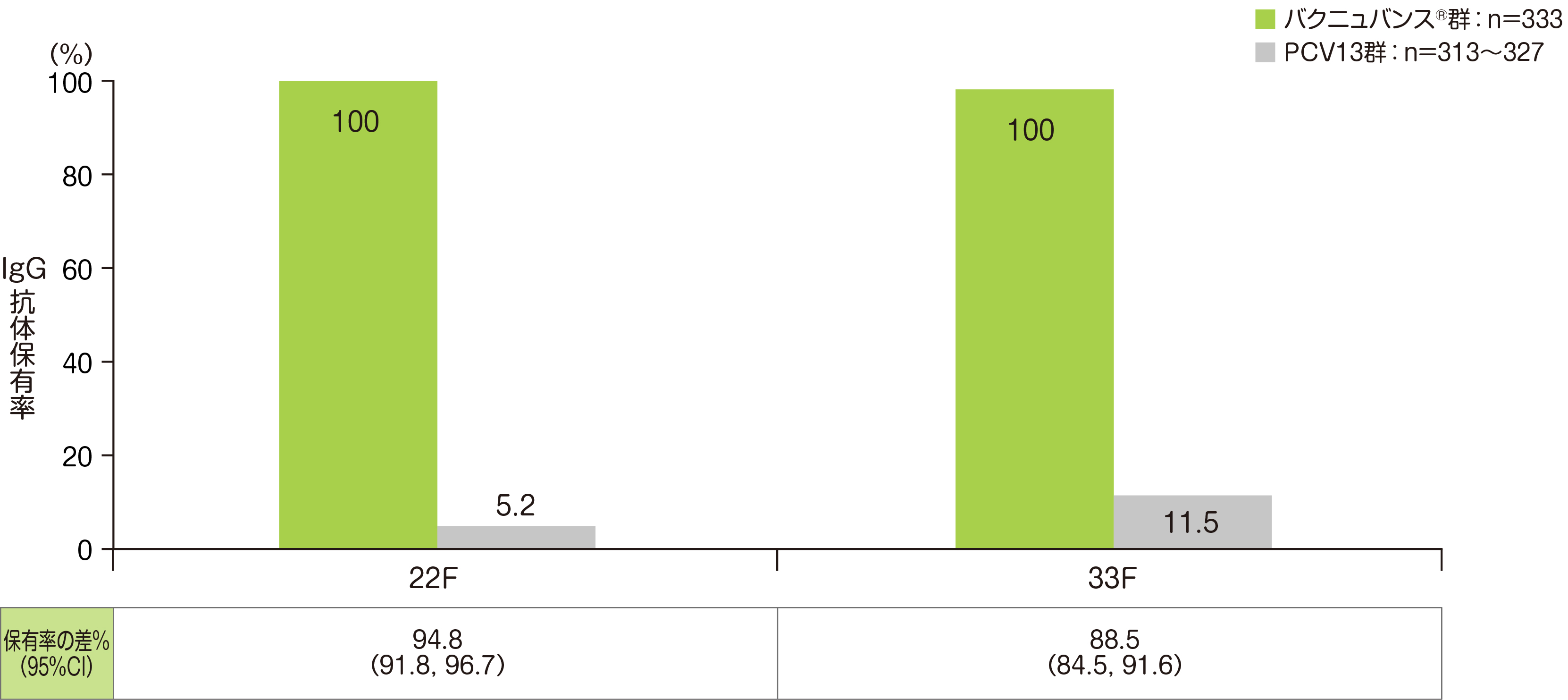
• PCV13群の血清型において解析可能な例数は異なる。治験実施計画書を逸脱していない全ての被験者(PP集団:バクニュバンス®群、PCV13群)を用い、本評価において、解析可能な例数を用いて集計を行った。
• Cochran-Mantel-Haenszel法による層別割合の差を算出し、Miettinen & Nurminen法(層別因子:月齢)で95%CIを算出した。
※ 血清型特異的IgG抗体保有率:血清型特異的IgG抗体濃度≧0.35μg/mLの被験者割合
安全性の主要評価項目である、「接種後14日までの事前に規定した注射部位および全身性の有害事象、試験終了までの重篤な副反応」では、安全性解析対象例(バクニュバンス®群347例、PCV13群346例)の内、接種後14日間の事前に規定した注射部位の有害事象はバクニュバンス®群で319例(91.9%)、PCV13群で316例(91.3%)が認められました。
接種後14日間の事前に規定した全身性の副反応(全ての時点の治験薬接種後)はバクニュバンス®群で263例(75.8%)、PCV13群で255例(73.7%)が認められました。
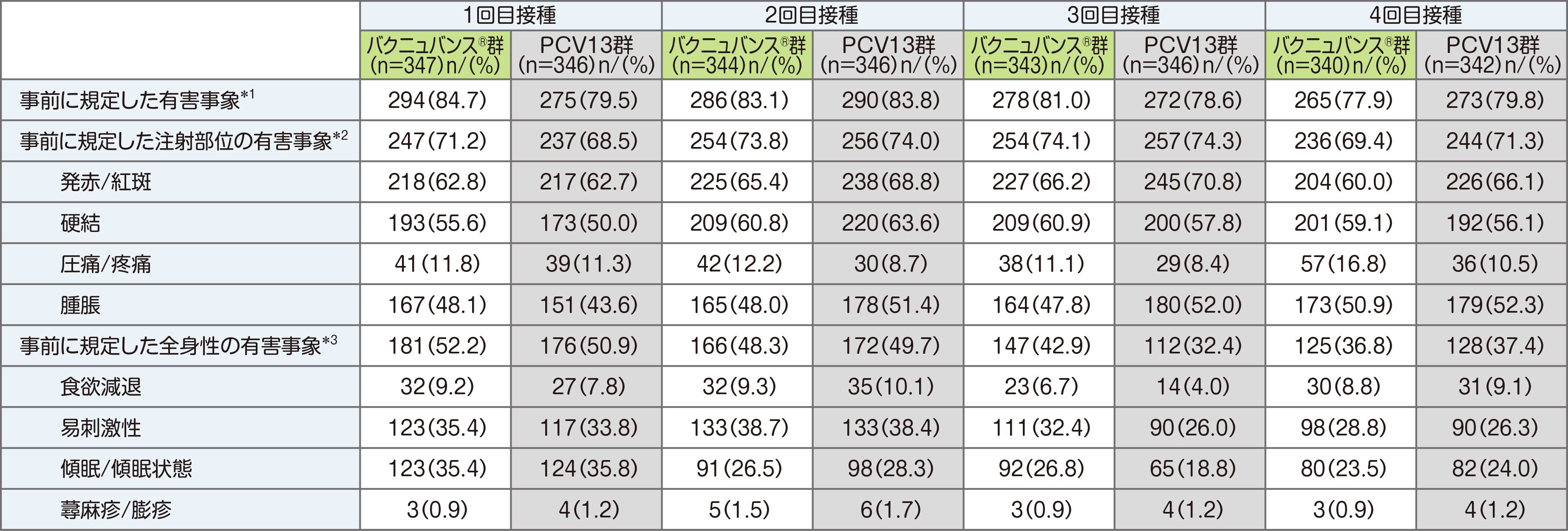
*1 事前に規定した注射部位の有害事象、事前に規定した全身性の有害事象のいずれかが1件以上発現した数
*2 発赤/紅斑、硬結、圧痛/疼痛、腫脹のいずれかが1件以上発現した数
*3 食欲減退、易刺激性、傾眠/傾眠状態、蕁麻疹/膨疹のいずれかが1件以上発現した数
重篤な副反応はバクニュバンス®群1例(痙攣発作)、PCV13群1例(発熱)に認められました。試験中止例は、バクニュバンス®群で9例(被験者の親・保護者の同意撤回8例、追跡不能1例)、PCV13群で6例(被験者の親・保護者の同意撤回6例)で、死亡例はバクニュバンス®群、PCV13群ともに認められませんでした。
バクニュバンス®接種後1日目から7日目における発熱に関しては、バクニュバンス®接種後の最高体温が38.0℃以上であった被験者割合は8.7%~19.4%でした。また、最高体温が40.0℃以上であった被験者割合は0%~1.5%でした。
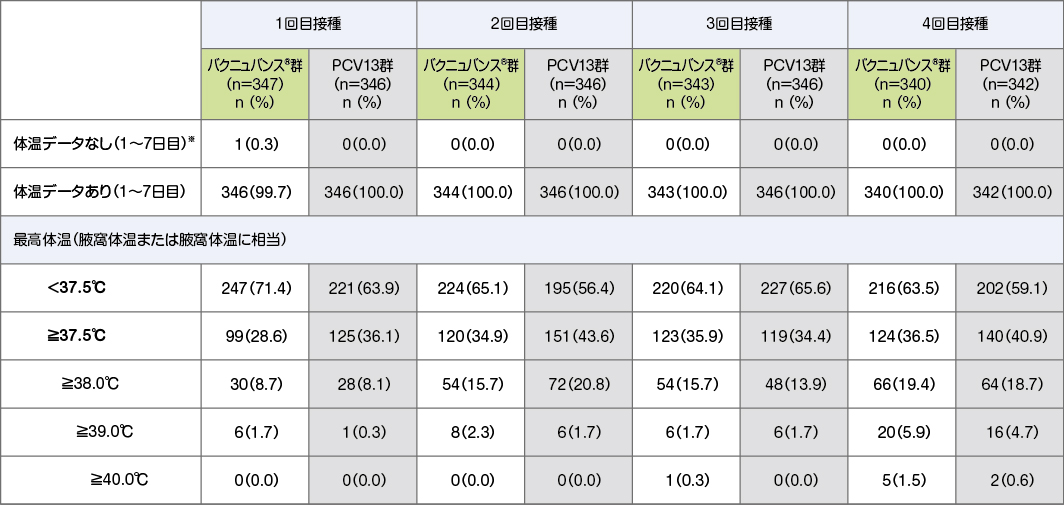
* 1~7日目までの体温測定が未報告、または腋窩体温に変換できなかった参加者を含む。
• 腋窩以外で体温測定した場合は、腋窩体温に換算した。
• 最高体温別の被験者割合は、体温データのある被験者数をもとに算出した。
• 一人の被験者が複数のカテゴリーに含まれることもある。
バクニュバンス®は、健康小児を対象とした臨床試験に加え、鎌状赤血球症の小児、HIV感染の小児、早産児、同種造血幹細胞移植を受けた小児など、ハイリスク小児を対象とした臨床試験も行われています。

※1 CD4+T細胞数が200個/μmol以上かつ血漿 中HIV RNA値≦50,000 copies/mLの6~17歳の小児を対象とした試験
※2 出生時妊娠37週未満
※3 本試験は、小児 (n=14) および成人人(n=260) で実施された。
1) 承認時評価資料: 海外第Ⅲ相試験(024試験)
2) Banniettis N, et al. Vaccine. 2022; 40(44):6315-6325.
3) 承認時評価資料: 海外第Ⅲ相試験(027試験)
4) Bili A, Dobson S, et al. Vaccine. 2023; 41(3): 657-665.
5) 承認時評価資料: 海外第Ⅲ相試験(029試験)
6) Lupinacci R, et al. Vaccine. 2023;41(5): 1142-1152.
7) 承認時評価資料: 海外第Ⅲ相試験(031試験)
8) 承認時評価資料: 国内第Ⅲ相試験(033試験)
9) 承認時評価資料: 海外第Ⅲ相試験(023試験)
10) Quinn CT, et al. Blood Adv. 2023; 7(3): 414-421.
11) 承認時評価資料: 海外第Ⅲ相試験(030試験)
12) Wilck M, et al. AIDS. 2023;37(8):1227-1237
13) 承認時評価資料: 海外第Ⅲ相試験(022試験)

本動画では、小児侵襲性肺炎球菌感染症予防の観点から、0歳児における初回免疫での免疫獲得の重要性について、詳しくご説明いたします。


バクニュバンス®は、新たに侵襲性の高い22Fと33Fの血清型を追加した、沈降15価肺炎球菌結合型ワクチンです。本動画では、小児肺炎球菌感染症に関する血清型におけ […]
WEB講演会にアクセスし「このページは閲覧を制限しています」と表示された方は こちら>>
このサイトでは、医療用医薬品を適正にご使用いただくため、医師、歯科医師及び薬剤師などの医療関係者の方を対象に、主としてMSD株式会社の医療用医薬品に関する情報を提供しています。
下記の「はい」をクリックした場合、「MSD Connect ご利用規約」及び「ウェブサイトのご利用条件」を理解したうえで、内容に同意したものとみなします。
2024年11月にご利用規約を改訂致しました。上記リンクよりご確認ください。
あなたは医療関係者ですか?